 As
propriedades termodinâmicas calculadas com os parâmetros
do campo de força CVFF estão listadas na Tabela.
Enquanto o valor de DHvap
concorda muito bem com o valor experimental, a densidade
calculada é subestimada. Isto provavelmente é função
da ausência de efeitos de polarização, já
que as cargas do campo de força CVFF fornecem um momento
dipolo clássico médio para uma molécula de
octanol em fase líquida muito inferior ao experimental
(Tabela). Como o momento dipolo experimental do octanol em fase gás
é 1,7 D, as cargas do campo de força CVFF não
descrevem adequadamente o comportamento eletrostático em
solução. Para incluir efeitos de polarização,
utilizamos nas simulações em fase líquida as
cargas CHELPG (Charges from Electrostatic Potential Grid-based)
da conformação all-trans do octanol obtidas em
nível Hartree Fock usando uma base
As
propriedades termodinâmicas calculadas com os parâmetros
do campo de força CVFF estão listadas na Tabela.
Enquanto o valor de DHvap
concorda muito bem com o valor experimental, a densidade
calculada é subestimada. Isto provavelmente é função
da ausência de efeitos de polarização, já
que as cargas do campo de força CVFF fornecem um momento
dipolo clássico médio para uma molécula de
octanol em fase líquida muito inferior ao experimental
(Tabela). Como o momento dipolo experimental do octanol em fase gás
é 1,7 D, as cargas do campo de força CVFF não
descrevem adequadamente o comportamento eletrostático em
solução. Para incluir efeitos de polarização,
utilizamos nas simulações em fase líquida as
cargas CHELPG (Charges from Electrostatic Potential Grid-based)
da conformação all-trans do octanol obtidas em
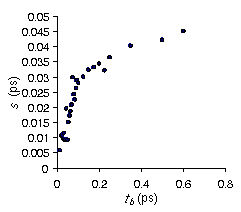
nível Hartree Fock usando uma base 6-31G**. Como esperado, as cargas CHELPG forneceram um momento dipolo maior que o momento dipolo experimental em fase gás, porém menor que o experimental em fase líquida. Embora com as cargas CHELPG a densidade calculada se aproxime do valor experimental, o DHvap é muito endotérmico. Como a cadeia do octano é muito flexível e possui um grupamento hidroxila que afeta fortemente o potencial eletrostático molecular (MEP), as cargas derivadas para a conformação
all-trans não devem reproduzir o MEP correto das inúmeras conformações assumidas pelo octanol em fase líquida. Desta forma, a média temporal das cargas CHELPG de todas as conformações da molécula do 1-octanol deveria reproduzir melhor o MEP de cada conformação. Contudo, o grande número de conformações impede o cálculo do MEP e consequentemente as cargas CHELPG de cada conformação. Felizmente, conformações obtidas entre passos de MD menores do que o tempo de correlação t estão tão correlacionadas que não fornecem informação estatística nova. No presente trabalho, assumimos que conformações obtidas após 2t passos de MD estão descorrelacionadas. Um procedimento eficiente na obtenção do tempo de correlação é a aplicação da ineficiência estatística (s). Para um processo Markoviano, o tempo de correlação e a ineficiência estatística são relacionados por s ( 2t. Com isto, calculamos a ineficiência estatística do ângulo de torção do grupo hidroxila (Figura), já que o mesmo possui uma grande influência no MEP e consequentemente nas cargas de cada conformação. Em seguida, selecionamos 50 estruturas do octanol com conformações descorrelacionas para o grupo hidroxila. Obtivemos a média temporal das cargas CHELPG das conformações selecionadas e calculamos as propriedades densidade e DHvap. Embora estas propriedades não estejam em concordância com os valores experimentais, os números calculados são melhores do que os obtidos com as cargas CHELPG da conformação all-trans. Além disto, o momento de dipolo calculado para a fase líquida comparado com o experimental sugere que a média temporal das cargas CHELPG dão uma melhor descrição do comportamento eletrostático do líquido. Estes resultados suportam nossa metodologia para derivar cargas parciais para moléculas flexíveis.