CARGAS ATÔMICAS EM COMPOSTOS INORGÂNICOS: UM ESTUDO SISTEMÁTICO PARA A SUA OBTENÇÃO.
Kátia J. de Almeida (IC),Willian R. Rocha (PG) e Wagner B. De Almeida (PQ)
Laboratório de Química Computacional e Modelagem Molecular (LQC-MM)
Departamento de Química, ICEx, UFMG, Belo Horizonte-MG, Brasil.
Palavras-chave: cargas atômicas, compostos inorgânicos, ab-initio.
O conceito de cargas atômicas em moléculas, apesar de sua natureza arbitrária, é parte integrante do pensamento químico. As cargas atômicas encontram aplicações em diversos estudos1. Métodos teóricos para a obtenção dessa propriedade são de grande utilidade, visto que sua determinação experimental em moléculas é indireta, geralmente difícil e se limita a cristais. A inexistência de um operador quanto-mecânico, associado à cargas atômicas, faz com que não exista um único método para a sua obtenção. Logo, várias metodologias teóricas1 para modelos de cargas através da função de onda, obtida por métodos ab initio, são encontradas na literatura. A comparação dos valores absolutos de cargas calculados segundo diferentes métodos necessita de significado, contudo certos critérios mínimos são desejaveis para que um conjunto de cargas atômicas sejam aceitáveis, ou seja, os valores obtidos devem possuir um interpretação física clara, sendo portanto compatíveis com a intuíção química.
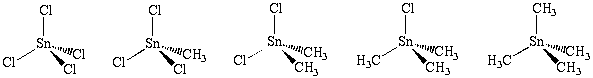
Apesar destes métodos serem extensivamente utilizados com êxito na predição da reatividade de compostos orgânicos, um estudo sistemático acerca da aplicabilidade dos mesmos em moléculas inorgânicas, pelo que sabemos, ainda não foi realizado. Dando continuidade aos nossos estudos em simulação de líquidos inorgânicos e na reatividade de compostos organoestânicos2, neste trabalho nós investigamos a performance e aplicabilidade de diferentes metodologias teóricas na descrição das cargas atômicas dos compostos organoestânicos mostrados no esquema abaixo.
Os cálculos de otimização de estrutura e obtenção de cargas foram feitos ao nível Hartree-Fock de teoria, com os efeitos de correlação eletrônica sendo tratados utilizando-se teoria de perturbação em segunda ordem (MP2). Para os compostos mostrados no esquema 1, a função de base 6-31G(d,p) foi utilizada para os átomos de C, H e Cl. Para o átomo de Sn, as funções de base 3-21G, DZVP e pseudopotencias relativísticos com funções de base de qualidade duplo-z para os elétrons de valência foram utilizadas. As cargas atômicas foram avaliadas através de metodologias bem estabelecidas e frequentemente utililizadas, como os métodos ChelpG, NPA, Mulliken e AIM. Tal procedimento foi efetuado com o objetivo de avaliar os efeitos de diferentes funções de base e a utilização de pseudopotenciais sobre o átomo de estanho, bem como efeitos de correlação eletrônica na qualidade das cargas atômicas fornecidas por estes métodos. No caso específico do método ChelpG, que tem uma dependência explícita com o raio de van der Waals(RvdW) em seu formalismo, um estudo minuncioso da dependência destas cargas com a variação do RvdW para o átomo de Sn foi realizado, uma vez que, para a maioria dos compostos inorgânicos não existe um RvdW bem definido para o átomo metálico.
Alguns resultados obtidos são sumarizados na tabela 1. Como pode ser visto, o conjunto de cargas obtidos pelos métodos de Mulliken e NPA não forneceram uma reprodução coerente do comportamento químico esperado, ou seja, ao substituirmos os átomos de Cl por grupos metila, a tendência esperada seria que a densidade eletrônica sobre o átomo de Sn, diminuísse progressivamente. O comportamento destes métodos foi invariante quanto à mudança de funções de base e nível de cálculo. As cargas pelo método ChelpG, mostraram ter uma dependência pronunciada com relação ao RvdW, funções de base, nível de cálculo e utilização de pseudopotenciais utilizados para o átomo de Sn. Foi verificado a existência de um limite ótimo de RvdW para o átomo metálico, onde as cargas obtidas fornecem um comportamento esperado para a reatividade destes compostos organoestânicos. Dentre os métodos utilizados, aquele baseado na topologia da densidade eletrônica, AIM, foi o que forneceu melhores resultados e menor sensibilidade quanto às mudanças de funções de base, nível de cálculo e efeitos de correlação eletrônica. Uma discussão mais detalhada de todos estes métodos, mostrando a sensibilidade dos mesmos com relação aos substituintes, uso de diferentes funções de base e o uso de diferentes níveis de cálculo será abordado, demonstrando a aplicabilidade dos mesmos na obtenção de cargas atômicas em moléculas inorgânicas.
Tabela 1 – Valores de cargas atômicas para o átomo de Sn.
|
|
qSn |
|||
|
|
Mulliken |
NPA |
ChelpG* |
AIM |
|
SnCl4 |
0,848 |
1,997 |
1,399 |
2,140 |
|
SnCl3Me |
0,871 |
2,037 |
1,376 |
2,089 |
|
SnCl2Me2 |
0,893 |
2,069 |
1,355 |
2,050 |
|
SnClMe3 |
0,917 |
2,079 |
1,300 |
2,012 |
|
SnMe4 |
0,938 |
2,049 |
1,203 |
1,970 |
* utilizando um RvdW de 2,17 para o Sn.
Referências:
[1] P.H.Guadagnini, A.A.de Souza e R.E.Bruns,Química Nova,19,148,(1996).
[2] (a) K. J. de Almeida, W. R. Rocha and W. B. De Almeida, Chem. Phys. Lett. 2000 no prelo. (b) W. R. Rocha and W. B. De Almeida, Organometallics, 17, 1961, (1998). (c) W. R. Rocha and Wagner B. De Almeida, Int. J. Quantum Chem., 65, 643, (1997).
FAPEMIG, CNPq, PADCT, CENAPAD-MG/CO