
PROPRIEDADES ELETRÔNICAS DE HEXILTIOFENOS ANALISADAS POR MÉTODOS AB INITIO E PELA TEORIA DO FUNCIONAL DE DENSIDADE
MARCOS ANTÔNIO DE OLIVEIRA(PG)a, EDER SEVERINO XAVIER(IC)a, JEAN-MICHEL PERNAUT(PQ)b, WAGNER BATISTA DE ALMEIDA(PQ)a.
aLaboratório de Química Computacional e Modelagem Molecular (LQC-MM)
bLaboratório de Novos Materiais
Departamento de Química-ICEx,
Universidade Federal de Minas Gerais
Palavras-chave: hexiltiofenos, Hartree-Fock, teoria-do-funcional-de-densidade.
0s alquiltiofenos além de ter as propriedades eletrônicas dos polímeros não substituídos, são processáveis, assim é possível efetuar a síntese das moléculas e a seguir testá-las como semicondutores, enquanto os derivados não substituídos necessitam de um precussor processável. Os alquiltiofenos como os hexiltiofenos (fig. 1, com n=6), têm sido utilizados na construção de diodos emissores de luz, com o comprimento de onda da emissão controlado através do tamanho da cadeia lateral do grupo alquila substituinte.
Figura 1 Molécula de poli(alquiltiofeno).
Pretendemos estabelecer relações claras de condutividade entre olígômeros derivados dos hexiltiofenos, com um número crescente de unidades monoméricas, para corroborar resultados experimentais e também para servir de guia para químicos sintéticos e cientistas de materiais, que tenham aplicações práticas na microeletrônica. Entretanto, a execução de cálculos apropriados no nível Ab Initio, com correlação eletrônica, dificultaria nossas tarefas, por isso optamos por calcular as geometrias no nível HF (Hartree-Fock) e posteriormente utilizar essa geometria para cálculos correlacionados usando a DFT (Teoria do Funcional de Densidade), nas aproximações local, GGA (aproximação do gradiente generalizado) e híbrida (o funcional de troca é oriundo do método de HF)1. A aproximação local foi dada pelo funcional S-VWN, a GGA pelo funcional B-LYP e a híbrida pelos funcionais B3P83 e B3LYP1. Para realizar nossos objetivos tomamos a lacuna de energia (banda de energia proibida) entre as bandas de condução e de valência como o valor em módulo da diferença entre os valores da afinidade eletrônica (AE = -ELUMO) e o potencial de ionização (PI = -EHOMO). Os valores calculados por nós da largura da lacuna de energia (entre as bandas de condução e de valência) são uma medida da condução no polímero condutor, e esses valores são normalmente apresentados como uma função do inverso do número de unidades monoméricas da cadeia. O método HF sempre superestima essa propriedade, acontecendo o mesmo com os métodos híbridos da DFT (B3-LYP e B3-P86), devido à contribuição de troca do método de HF. Em contrapartida os funcionais local (S-VWN) e GGA (B-LYP) apresentam resultados comparáveis aos experimentais para derivados não substituídos do tiofeno. É possível verificar que existe uma variação dos valores da lacuna de energia com o número de monômeros, demonstrando que os oligômeros com número par de unidades monoméricas são melhores condutores que as formas com número ímpar de unidades, fig.2.
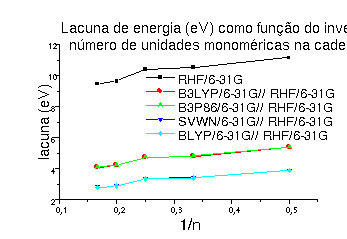
Figura 2 Lacuna de energia entre as bandas de condução e de valência, responsável pelas propriedades semicondutoras dos monômeros.
Conclusões: É possível afirmar com base nas geometrias calculadas que quando o número de unidades monoméricas é ímpar a condução linear é facilitada pela proximidade de duas unidades em planos tendo como obstáculo para a condução apenas uma outra unidade em um plano diferente. A DFT, nas suas aproximações local e GGA, têm demonstrado excepcional aplicação para semicondutores orgânicos.
Bibliografia
1-Análise de propriedades eletrônicas de derivados do a,a'-tiofeno
M. A. de Oliveira, J.-M. Pernaut, W.B. de Almeida.
XIII Encontro Regional da SBQ-MG, São João Del Rei , de 03 a 05/11/99.
(CNPq, CENAPAD-MG/CO, FAPEMIG).