A LACUNA DE ENERGIA DO P-FENILTIOFENO COMO FUNÇÃO DA CONDUTIVIDADE POR MÉTODOS AB INITIO E PELA TEORIA DO FUNCIONAL DE DENSIDADE
MARCOS ANTÔNIO DE OLIVEIRA(PG)a, JEAN-MICHEL PERNAUT(PQ)b, WAGNER BATISTA DE ALMEIDA(PQ)a.
aLaboratório de Química Computacional e Modelagem Molecular (LQC-MM)
bLaboratório de Novos Materiais
Departamento de Química-ICEx,
Universidade Federal de Minas Gerais
Palavras-chave: p-fenil(r-tiofeno), capacitores, teoria-do-funcional-de-densidade.
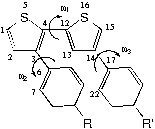
O p-feniltiofeno) (fig. 1), têm potenciais aplicações para a construção de dispositivos eletrônicos, especificamente para capacitores, sendo por esse motivo um protótipo adequado para o estudo das propriedades moleculares eletrônicas, que
Figura 1 O p-feniltiofeno pode ter as posições para dos anéis fenila substituídas por grupos aceitadores ou doadores de elétrons, permitindo variar a lacuna de energia entre as bandas de condução e de valência.
gerenciam a condução elétrica em semicondutores orgânicos devido à possíveis substituições por grupos eletroativos na posição para dos anéis fenila1. Trabalhamos anteriormente com este sistema para testar as conformações não substituídas nos seus mínimos de energia e estados de transição e comparar com um potencial, também gerado por nós, baseado em um ajuste de séries de Fourier truncadas2.
Agora temos o interesse na obtenção de resultados teóricos que corroborem experimentos de espectroscopia de voltametria cíclica e de ultra-violeta (medidas iniciadas no nosso departamento), e deste modo prever a lacuna de energia de semicondutores orgânicos, o que tem nos levado a utilizar cálculos correlacionados de alto nível. Cálculos Ab Initio HF (Hartree-Fock) e cálculos com correlação eletrônica, baseados na DFT (Teoria do Funcional de Densidade), nas aproximações local (S-VWN), GGA (Aproximação do Gradiente Generalizado no funcional B-LYP) e híbrida (a contribuição de troca provem do método HF, no funcional B3-LYP e B3-P86) foram realizados. Os resultados mostraram que a energia dos orbitais de fronteira é suficiente para descrever o ganho e a perda de elétrons, sendo o formalismo exato na DFT, sustentado pelo teorema de Janak. Assim as energias dos orbitais de fronteira descrevem o potencial de ionização e a afinidade eletrônica. Na figura 2 torna-se perceptível o fato de que embora as conformações tenham energias bastante próximas2 é possível estabelecer diferenças de condutividade baseadas nos valores das lacunas de energia. A posterior substituição dos grupos R e R' da molécula de p-feniltiofeno possibilitará o controle da condutividade do material em aplicações práticas.
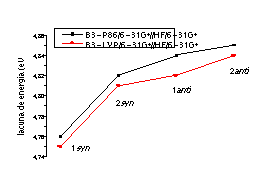
Figura 2 As conformações syn são mais estáveis do que as conformações anti, devido à interações mais efetivas entre os grupos fenila, que esmaecem no caso das moléculas de conformação anti.
Na tabela 1 podemos comparar resultados para outras aproximações da DFT.
Tabela 1 Valores para lacuna de energia (eV) calculadas nas aproximações local
(S-VWN), GGA (B-LYP) e híbrida (B3-P86 e B3-LYP), geometria RHF/6-31G*.
|
Conformação |
S-VWN |
B-LYP |
B3-P86 |
B3-LYP |
|
1-syn |
3,39 |
3,38 |
4,76 |
4,75 |
|
1-anti |
3,45 |
3,44 |
4,84 |
4,82 |
Conclusão: É um fato conhecido, que os valores calculados pelos métodos híbridos são superestimados, assim como àqueles dos obtidos através dos métodos ab initio, mas as aproximações local e GGA mostram resultados próximos aos experimentais, com diferenças de menos de 1 eV.
Bibliografia:
1-A density functional, Ab Initio and Semi-empirical study about Torsional Potential of Oligo-Phenylthiophenes
M. A. de Oliveira, H. A. Duarte, J.-M. Pernaut, W.B. de Almeida. Submetido para publicação.
2-Análise de propriedades eletrônicas de derivados do a,a'-tiofeno
M. A. de Oliveira, J.-M. Pernaut, W.B. de Almeida.
XIII Encontro Regional da SBQ-MG, São João Del Rei , de 03 a 05/11/99.
(CNPq, CENAPAD-MG/CO, FAPEMIG).