PROPRIEDADES ELETRÔNICAS DE a,a'-TIOFENOS PELA TEORIA DO FUNCIONAL DE DENSIDADE
MARCOS ANTÔNIO DE OLIVEIRA(PG)a, HÉLIO ANDERSON DUARTE(PQ)a , ANA PAULA LAMOUNIER (IC)a, JEAN-MICHEL PERNAUT(PQ)b, WAGNER BATISTA DE ALMEIDA(PQ)a.
aLaboratório de Química Computacional e Modelagem Molecular (LQC-MM)
bLaboratório de Novos Materiais
Departamento de Química-ICEx
Universidade Federal de Minas Gerais
Palavras-chave: a,a'-tiofenos, funções-de-base, teoria-do-funcional-de-densidade.
Os derivados não substituídos do a,a'-tiofenos (fig. 1), com suas potenciais aplicações em sistemas eletrônicos são os protótipos de partida para a construção e entendimento de moléculas novas. Como a DFT (Teoria do Funcional de Densidade) têm produzido resultados para estes compostos em boa comparação com os resultados experimentais1, resolvemos investigar a natureza das informações produzidas por estes métodos, com mais rigor, otimizando as estruturas (obtidas inicialmente no nível HF, Hartree-Fock), no nível correlacionado da DFT, com variações nos conjuntos de funções de base utilizados. A partir desse ponto avaliamos a lacuna de energia entre as bandas de condução e de valência, aproximadas como a diferença de energia entre os orbitais de fronteira HOMO e LUMO1. As aproximações da DFT disponíveis foram utilizadas: local, GGA (aproximação do gradiente generalizado) e híbrida
Figura 1 Molécula de politiofeno.
(o funcional de troca inclui parte da energia de troca exata dada pelo método de HF). A aproximação local foi dada pelo funcional SVWN, a GGA pelo funcional BLYP e a híbrida pelos funcionais B3P86 e B3LYP. Os valores calculados da lacuna de energia não apresentaram mudanças qualitativas em função do inverso de número de monômeros na cadeia, fig 2, para resultados da DFT, com geometria RHF e também com as geometrias DFT otimizadas. Entretanto se observarmos quantitativamente os resultados, veremos diferenças significativas em eV, que levarão à diferenças de condutividade importantes, tabela1. Ainda é importante observar que os funcionais S-VWN e B-LYP apresentam resultados praticamente idênticos fig. 2 e tab. 1.
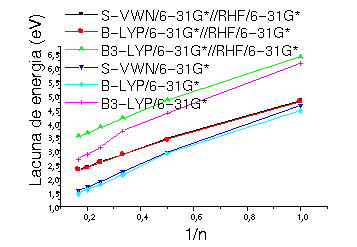
Figura 2 Valores da lacuna de energia calculados com a geometria RHF/6-31G* comparados a valores otimizados no nível DFT/6-31G*
Tabela 1 Valores calculados para a lacuna de energia com geometria RHF/6-31G*a e otimizados no nível da DFTb , também com o conjunto de funções de base 6-31G*.
|
Derivados |
S-VWNa |
B-LYPa |
B3-LYPa |
S-VWNb |
B-LYPb |
B3-LYPb |
|
t |
4,8 |
4,76 |
6,36 |
4,61 |
4,43 |
6,13 |
|
2t |
3,43 |
3,4 |
4,8 |
2,94 |
2,91 |
4,35 |
|
3t |
2,88 |
2,86 |
4,16 |
2,25 |
2,14 |
3,71 |
|
4t |
2,61 |
2,59 |
3,83 |
1,9 |
1,8 |
3,13 |
|
5t |
2,43 |
2,38 |
3,62 |
1,69 |
1,6 |
2,87 |
|
6t |
2,35 |
2,33 |
3,53 |
1,55 |
1,46 |
2,69 |
Conclusão:
Os valores calculados das lacunas de energia apresentam-se confiáveis na ausência de resultados experimentais, sendo então, uma ótima referência para os químicos sintéticos e cientistas de materiais interessados no sistema apresentado ou em outros similares.
Bibliografia
1-Análise de propriedades eletrônicas de derivados do a,a'-tiofeno
M. A. de Oliveira, J.-M. Pernaut, W.B. de Almeida.
XIII Encontro Regional da SBQ-MG, São João Del Rei , de 03 a 05/11/99.
(CNPq, CENAPAD-MG/CO, FAPEMIG).