Modelagem Molecular das
1-Fenil 1,2,3,4 Tetrahidro b-Carbolinas:
Potenciais Agentes Antimicrobianos
João Bosco P. da Silva (PQ)1, Marcelo Zaldini Hernandes (PG)1, Elba Lúcia C. de Amorim (PQ)2, Simone A. Amorim (IC)2 e Cláudia Sampaio de A. Lima(PQ)3
1 Departamento de Química Fundamental, 2 Departamento de Farmácia e
3 Departamento de Biofísica
Universidade Federal de Pernambuco - Recife (PE) 50740-250
Palavras-chaves b-carbolinas, AM1, agentes antimicrobianos
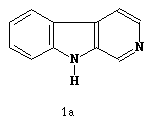
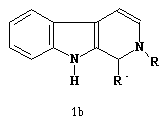
Uma classe de compostos que tem despertado bastante interesse nas últimas décadas devido suas propriedades farmacológicas são os alcalóides. O núcleo b-carbolina, Figura 1a, encontra-se presente no esqueleto de vários alcalóides indólicos, assim, como sua forma reduzida, o 1,2,3,4-tetrahidro-b-carbolina, Figura 1b, ambas apresentando atividades antitumorais e hipotensoras [1,2].
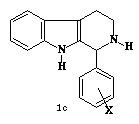
X
= 4-Cl, 4-F, 2-F, 3-CH3, 2,4-diOCH3,
4-N(CH3)2, 4-NHCOCH3,
2,6-diCl. 2,4-di-Cl
Figura 1. a) b-carbolina, b) tetrahidro-b-carbolina, c) 1-Fenil-b-carbolina.
Recentemente, a série das 1-Fenil-1,2,3,4-b-Carbolinas, Figura 1c, foi sintetizada e caracterizada espectroscopicamente por técnicas de infravermelho (IV), espectroscopia de massa e ressonância magnética nuclear (RMN) de próton (1H) e carbono 13 (13C) pelo nosso grupo. Inicialmente, esta classe de compostos foi submetida a testes preliminares de atividade contra vários microorganismos no Departamento de Antibióticos da UFPE. Os compostos testados apresentaram atividade significativa para representantes Gram-positivos, Gram-negativos, Álcool-ácido resistentes Leveduras e Fungos filamentosos.
Uma das hipóteses básicas com vistas ao desenvolvimento futuro de uma relação quantitativa entre a estrutura química e atividade biológica (QSAR) é a da existência de um conjunto de variáveis latentes que se correlacionam com a atividade da droga no sistema biológico [3]. Uma vez que não conhecemos qual ou quais destas variáveis são as mais importantes, o bom senso sugere que devemos tomar o maior número possível de variáveis físico-químicas. Além dos dados espectroscópicos, uma outra fonte de variáveis físico-químicas que podem ser utilizadas são parâmetros energéticos, estruturais e eletrônicos obtidos de Cálculos de Orbitais Moleculares (O.M.). Entretanto, estes parâmetros são bastante dependente da metodologia de cálculo empregada, que inclui informações conformacionais e do meio ao qual o substrato está submetido. A descrição do meio condensado através de um modelo, em particular, água como solvente, pode ser importante para uma melhor representação da droga em meio fisiológico.
Assim, neste trabalho, realizamos cálculos semi-empirícos AM1, para a molécula da série das 1-Fenil-b-carbolinas, com X = H, em duas aproximações: i) o substrato isolado (para representação da fase gasosa) e ii) o substrato interagindo com algumas moléculas de água (representando a primeira esfera de hidratação).
Em particular, duas conformações que distinguem-se pelas posição sin ou anti do grupo fenil em relação ao par isolado de elétrons do nitrogênio piperidínico foram investigadas.
As estruturas hidratadas foram inicialmente geradas a partir da análise da superfície de potencial eletrostático do substrato, realizada através da utilização do código AGOA. A posição e orientação de cada molécula de água foi determinada para reproduzir os mínimos de energia na hiper-superfície de potencial. Este código, escrito em FORTRAN, foi desenvolvido e implementado no Laboratório de Química Teórica da UFPE.
Nossos resultados indicam que em fase gasosa a conformação anti é mais estável por 1,43 kcal. Entretanto, com a adição de uma, duas e três moléculas de água, essa diferença torna-se 1,71, 6,00 e 10,00 kcal, respectivamente. Estes resultados indicam que a medida que águas de hidratação são incorporadas ao nosso substrato, a forma anti torna-se mais estável.
Finalmente, a inclusão de mais moléculas de água e a aplicação de modelos contínuos de solvatação sobre a super-molécula (substrato + águas de hidratação) representará a próxima etapa no aprimoramento da metodologia para a aquisição dos parâmetros a serem utilizados nas analises de QSAR.
Referências:
1 - T. A. K. Al-Allaf, M. T. Ayoub, L. Rasham, J. Inorg. Biochem., 38, 27(1990).
2 - S. Wawzonek, G. E. Nelson, J. Org. Chem., 27, 1377(1962).
3 – L. Eriksson, E. Johansson, Chemo. And Intell. Lab. Systems, 34,1(1996).