SÍNTESE
DE NOVOS DERIVADOS DA CLASSE 1-H-PIRAZOLO
[3,4-b]PIRIDINA
ANÁLOGOS À AMODIAQUINA
Cláudio
Márcio Lourenço (IC), Heloísa de Mello (PG) e
Aurea Echevarria (PQ)
Departamento
de Química-ICE-Universidade Federal Rural do Rio de Janeiro
Palavras-chave:
Pirazolopiridina, descarboxilação,
amodiaquina
Muitos
derivados polissubstituídos do sistema heterocíclico
1H-pirazolo[3,4-b]piridina tem sido sintetizados e
testados quanto à diversas atividades biológicas como
vasodilatadora, hipoglicêmica, anti-inflamatória,
anti-pirética e analgésica.1
Em
trabalhos anteriores, nosso grupo de pesquisa relatou a síntese
e avaliação leishmanicida de vários compostos
derivados do sistema pirazolopiridina,2,3 sendo que estes
compostos apresentavam um grupo éster carboxílico no
anel piridínico aril-substituído. Devido ao fato destes
compostos terem apresentado significativa atividade anti-parasitária
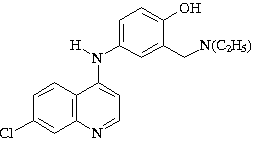
e ainda possuírem analogia estrutural com o antimalarial
amodiaquina, nosso interesse na busca de substâncias mais
ativas, levou-nos à obtenção de novos derivados
a partir de aminas heterocíclicas não aromáticas
e dos derivados (anteriormente sintetizados) descarboxilados.
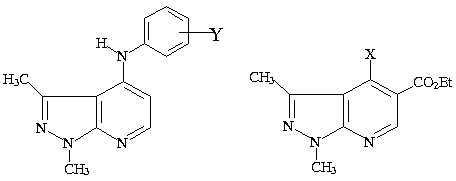
Na
presente comunicação relatamos a síntese de 4
novos derivados da série 4-(4’-X) –1H –
pirazolo[3,4-b]piridinas (1a–d) onde X=
piperidinil (1a), morfolinil (1b), pirrolidinil (1c)
e cicloexilaminil (1d); 5 novos da série 4-[3’ou
4’-X-fenilamino]-1H-pirazolo[3,4-b]-piridinas
(2a-e), onde Y= H (2a), p-CH3 (2b),
p-OCH3 (2c), p-OH (2d) e m-F
(2e).
Amodiaquina 2a - e 1a –d
Os
compostos intermediários foram sintetizados através de
reação de acoplamento entre o derivado
5-carboetoxi-4-cloro-(1’,3’)-dimetil-1H-pirazolo[3,4-b]piridina
e as aminas correspondentes. No caso das aminas não
aromáticas, foi utilizado etanol como solvente, em condições
de refluxo por 2 horas e, para as aminas aromáticas a reação
de acoplamento foi realizada em etileno glicol por 4 horas a
temperatura de refluxo. O intermediário clorado foi obtido à
partir de 1,3-dimetil-5-aminopirazol e b-amino-crotonitrila,
seguido por ciclização e cloração com
POCl3.
Os
derivados descarboxilados foram obtidos através de 3
metodologias diferentes:
Método
1: Os derivados ésteres foram submetidos inicialmente à
hidrólise alcalina para a obtenção de seus
respectivos ácidos correspondentes, sendo estes últimos
refluxados com DowTherm durante 4 horas a 2500C, com
posterior precipitação do produto em éter de
petróleo. Método 2: Os derivados ácidos
foram refluxados em ácido fosfórico concentrado durante
15 horas a 1700C, sendo então precipitados em
solução concentrada de hidróxido de amônio
e lavados com água destilada. Método 3: Os
ésteres foram diretamente refluxados em ácido
fosfórico, procedendo-se da mesma forma descrita no Método
2. Os produtos obtidos foram posteriormente recristalizados em
etanol. Os derivados 1a-c foram purificados em coluna de
sílica gel com diclorometano como eluente.
A
Tabela 1 mostra os resultados obtidos para as reações
de acoplamento fornecendo novos derivados 1a-d e os métodos
de descarboxilação dos derivados pirazolopiridinas
anilino-substituídos na forma de ésteres para dar
origem aos compostos 2a –e.
Tabela
1: Resultados obtidos
|
Composto
|
Rendimento
(%)
|
Composto
|
Rendimento
(%)
|
P.F
(°c)
|
|
1a
|
90
|
2a
|
43***
|
84-86
|
|
1b
|
60
|
2b
|
40*,60**
|
56-60
|
|
1c
|
67
|
2c
|
44***
|
259-260
|
|
1d
|
10
|
2d
|
51*
|
268-270
|
|
|
|
2e
|
24*
|
76-79
|
*Método
1,** Método 2, *** Método 3
De
acordo com os resultados obtidos, os dois métodos em que se
utilizou ácido fosfórico concentrado, foram mais
satisfatórios não só em termos de rendimento,
como também na facilidade de isolamento e purificação
dos compostos sintetizados. O melhor rendimento obtido correspondeu
aos métodos em que se utilizou ácido fosfórico
pode ser atribuído provavelmente à quebra da ligação
hidrogênio intramolecular com o hidrogênio ligado ao N
anilinico, fato este, que não ocorre em solventes apolares
como o Dow Therm.
A
descarboxilação dos ácidos e ésteres
utilizados foi comprovada através de métodos
espectrométricos tradicionais, onde foram empregados IV e
RMN1H, sendo o desaparecimento da banda de absorção
intensa em torno de 1670 cm-1 (IV) atribuído à
remoção da carbonila, enquanto que o aparecimento de
dois dubletos em torno de 6 e 8 ppm (J = 6,0 Hz) no espectro de RMN
1H pode ser atribuído ao acoplamento entre os
prótons do anel piridínico, o que não foi
observado nos compostos precursores destes.
Em relação às reações de
acoplamento, os melhores rendimentos foram apresentados, quando
utilizadas aminas secundárias, isto provavelmente devido ao
maior poder nucleofílico das aminas secundárias sobre
as anilinas.
1Ahluwaia,
V.K.;Goyal, B. Synthetic Communications, 26, 7, 1341,
1996.
2Bernardino,
A.M.R.e col. Heterocyclic Communications, 2, 5, 415,
1996.
3Mello,
H e col.; Química Nova, 22, 1, 26, 1999.
CNPq/PIBIC/CAPES