elucidação estrutural de Uréias e tiouréias assimétricas através de RMN 1D/ 2D, modelagem molecular e efeito dos substituintes
Andressa Esteves1 (PG), Aurea Echevarria1 (PQ),
Maria da Graça Nascimento (PQ)2
1Departamento de Química da Universidade Federal Rural do Rio de Janeiro
2 Departamento de Química da Universidade Federal de Santa Catarina
Dados da literatura demonstram o interesse de muitos pesquisadores no estudo estrutural de uréias e tiouréias. Este grande interesse pela elucidação da estrutura desses compostos vêm sendo atribuído às suas ínumeras aplicações, como por exemplo as atividades fungicida, pesticida, anti-hipertensiva e anti-HIV.1, 2
Estudos de RMN de 1H em solventes de diferentes polaridades aliados a dados cristalográficos têm contribuído para a determinação da estrutura de ariluréias.3
Em 1973, Lepore e colaboradores realizaram estudos conformacionais de N-aril-N-alquiluréias em solução e, através de dados de RMN estabeleceram as estruturas e a posição de seus substituintes.4
Neste trabalho, é relatado um estudo a elucidação estrutural utilizando dados de RMN 1D e 2D assimétricas, bem como o efeito eletrônico de grupos substituintes e resultados de modelagem molecular.
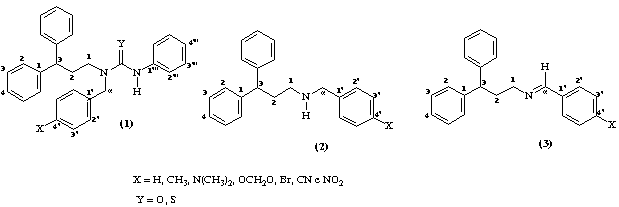
A síntese das N-[3,3-difenilpropil]-N-p-X-benzil-N’-feniluréias e tiouréias (1), foi realizada segundo relatado anteriormente.5 Os espectros de RMN foram obtidos em solução de CDCl3 a 200 e 50 MHz para 1H e 13C, respectivamente. Os dados de modelagem molecular foram obtidos utilizando o método semi-empírico AM1 através do programa MOPAC 6.0 para Windows 95.
A estratégia utilizada para a atribuição dos hidrogênios e carbonos nas moléculas das uréias e tiouréias envolveu inicialmente a atribuição dos espectros de RMN 1H e 13C das aminas precursoras, N-3,3-difenilpropil-N-p-X-benzilaminas (2) que por sua vez foram atribuídos em analogia às correspondentes Bases de Schiff (3). Os valores de deslocamento químico de RMN 1H para as aminas indicaram claramente a mudança da natureza do H, absorvendo na faixa 3,62 a 3,81, caracterizando o efeito de proteção do grupo amínico quando comparado ao imínico ((8,0). O sinal H-N carcterístico das aminas, apresentou-se na faixa 1,55 a 1,72.
Os valores dos deslocamentos químicos dos carbonos alquílicos e aromáticos ligados ao C3 das aminas mostraram-se semelhantes aos das correspondentes Bases de Schiff. Entretanto, para C1 e C3 observou-se maior efeito de blindagem devido ao NH. O C pôde ser assinalado inequivocamente na faixa de 48,0 (iminas 160,0). Os valores observados para os carbonos e hidrogênios do anel benzílico p-substituído foram coerentes com os efeitos eletrônicos dos substituintes.
O assinalamento dos hidrogênios nas moléculas das uréias e tiouréias apresentou-se similar às aminas correspondentes com exceção de H1 e H no caso das tiouréias, devido ao efeito do enxofre como átomo pesado. Experimentos bidimensionais de 1H x 1H-cosy mostraram as interações esperadas entre os hidrogênios da cadeia alquílica. No caso de uréia p-NO2 substituída observou-se o acoplamento de H com H3, enquanto que as tiouréias p-CH3 e OCH2O mostraram acoplamento entre H e H1. A absorção do N-H, apresentou forte desblindagem em relação às aminas correspondentes, 5,9 e 6,8 para as uréias e tiouréias, respectivamente.
A análise dos espectros de RMN 13C para as uréias e tiouréias foi realizada considerando-se as moléculas em três etapas. Na primeira etapa assinalou-se os carbonos da porção 3,3-difenilpropil por analogia às respectivas aminas, observando-se um efeito de proteção de aproximadamente 3ppm, para as uréias e desproteção de 1 ppm para as tiouréias, em função da posição do anel benzílico no espaço, conforme os resultados de modelagem molecular.
Na segunda etapa, foram atribuídos os carbonos da porção proveniente do isocianato e isotiocianato de fenila. As absorções observadas apresentaram-se na mesma faixa, para cada série sem variações significativas.
Comparando-se os deslocamentos químicos do C1’” para as duas séries, observaram-se menores valores para as tiouréias, provavelmente devido ao efeito do átomo de enxofre. Este efeito pôde ser ressaltado através da modelagem molecular da tiouréia não-substituída, indicando interação espacial do enxofre e o C1’”.
Os carbonos do anel benzílico absorveram em regiões coerentes com a natureza dos efeitos eletrônicos dos substituintes, tendo apresentado correlações significativas para C x F (uréias, r=0,93; tiouréias, r=0,94). Os C=O e C=S apresentaram 155,09 a 156,34 e 181,82 a 182,17, respectivamente, indicando também o efeito do átomo pesado para as tiouréias.
A metodologia utilizada, basicamente dados de RMN 1H, 13C e modelagem molecular, permitiu a caracterização correta das estruturas propostas para as uréias e tiouréias assimetricamente substituídas. CAPES/CNPq
1Debnath, A.K. J. Med, Chem.42,249,1999; 2Vajragupta, O. e col. J. Pharm. Sciences.85,3,258,1996; 3Wawer, I e col. Molecular Struct.344,251,1995; 4Lepore, G.; e col. M. J. Org. Chem.38,15,2590,1973; 5Souza, A.E. e col. 22a RA da SBQ, QO 149,1999.