BARREIRAS
ROTACIONAIS EM CARBAMATOS, DETERMINADAS POR CÁLCULOS
COMPUTACIONAIS
Ernani Abicht Basso (PQ), Paulo Roberto de
Oliveira (PG), Franciele Wiectzycoski (IC), Barbara Celânia
Fiorin (PET)
Departamento
de Química – Universidade Estadual de Maringá
(UEM)
Maringá
– PR
palavras-chave:
barreira rotacional, carbamatos, ab initio
Introdução
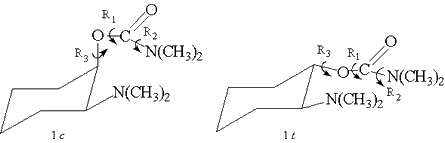
Os
derivados carbamatos tem o mais amplo espectro da atividade biológica
de quaisquer classe de compostos orgânicos. Estes compostos são
conhecidos como anticolinesterásicos por inibirem a ação
da acetilcolinesterase, que é a enzima responsável pela
inibição do neurotransmissor acetilcolina1.
Em estudos anteriores, em nossos laboratórios, foi sintetizada
a mistura cis (1c) e trans –
1-(((dimetilamino)carbonil)oxi)-2-N,N-dimetilaminocicloexano (1t),
a qual tem sido submetida a testes de atividade anticolinesterásica.
Figura
1: Representação das rotações R1,
R2, R3 nos compostos 1c e 1t .
Objetivos
Como
a ação farmacológica de qualquer composto está
diretamente associada ao seu arranjo espacial, os compostos em estudo
(1c e 1t ), foram investigados com respeito a variação
de energia associada a giros de 360o ( em intervalos de
100 ) para os ângulos torcionais das rotações
R1, R2 e R3 ( Figura 1), de forma a
identificar as energias relativas das conformações2.
Métodos
Os
compostos 1c e 1t foram otimizados pelos métodos
semi-empíricos (AM1, PM3 e MNDO) e Ab Initio (STO-3G e 3-21G).
As estruturas obtidas da otimização por cada um destes
métodos foram submetidas aos giros definidos na Figura 1. Os
cálculos foram realizados com o programa GAUSSIAN-943.
Resultados
Os
ângulos torsionais obtidos pelos diferentes métodos
computacionais são apresentados na Tabela 1 e representam o
ângulo mais estável para cada rotação (R1,
R2 e R3).
Tabela
1. Ângulos torsionais mais estáveis para os compostos 1c
e 1t determinados por métodos semi-empíricos
( AM1, PM3, MNDO ) e ab initio ( STO-3G e 3-21G ).
|
Composto
|
Rotações
|
AM1
|
PM3
|
MNDO
|
STO-3G
|
3-21G
|
|
|
R1
|
183
|
182
|
178
|
177
|
193
|
|
1
c
|
R2
|
16
|
14
|
1
|
29
|
4
|
|
|
R3
|
113
|
122
|
124
|
105
|
100
|
|
|
R1
|
164
|
175
|
173
|
175
|
166
|
|
1
t
|
R2
|
170
|
180
|
176
|
193
|
178
|
|
|
R3
|
243
|
242
|
238
|
260
|
264
|
Conclusão
Os
ângulos torsionais obtidos pelos métodos computacionais
apresentados na Tabela 1 embora diferentes quantitativamente, não
apresentam diferenças significativas qualitativamente, o que
significa que todos estes ângulos se encontram próximos
da região de energia mínima para cada rotação.
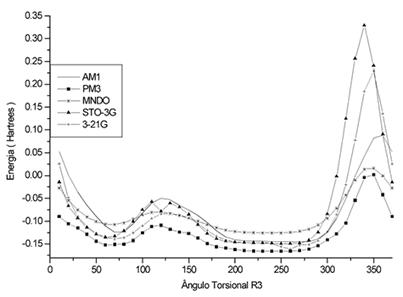
Isto pode ser confirmado, por exemplo, analisando a Figura 1
referente a variação de energia para rotação
R3 do composto 1t. A Figura 1 demonstra também
a conformação mais estável do referido composto.
Esta análise foi realizada para cada uma rotações
dos compostos 1c e 1t.
Bibliografia
Gilman,
A.G.; Roll, T. N.; Nies, A. S.; Taylor, P. .As Bases Farmacológicas
Da Terapêutica, 8a Ed. , Guanabara Koogan
Rio de Janeiro , RJ ( 1991).
Silverman,
R. B.. “The Organic Chemistry of Drug design and Drug Action”,
Academic Press, London, 1992.
Foresman,
A.B.; Frisch, A.; “Exploring Chemistry with Electronic
Structure Methods”, Second
Edition, 1995 – 1996 .Gaussian, Inc.Pittsburgh, PA
U.S.A. Capes