PLANEJAMENTO, SÍNTESE E ATIVIDADE ANTIINFLAMATÓRIA DE NOVAS SULFONILIDRAZONAS FENILACÉTICAS
Juliana Bossan de Velasco (IC)1, Emanuel Gonçalves Amarante (IC)1, Lis Helena Pinheiro Teixeira (PG)1,2, Ana Luisa Palhares Miranda (PQ)1, Carlos Alberto Manssour Fraga (PQ)1 & Eliezer Jesus Barreiro (PQ)1
1LASSBio – Departamento de Fármacos – Faculdade de Farmácia – UFRJ
2Departamento de Química Orgânica – Instituto de Química – UFRJ
palavras-chave: antiinflamatório; sulfonilidrazônicos; inibidor enzimático
INTRODUÇÃO: A inflamação é considerada como a resposta do organismo a uma agressão tecidual, geralmente acompanhada de sinais clínicos como calor, dor, edema, tumor e perda de função1. Os agentes antiinflamatórios não esteroidais (AINEs) são os medicamentos mais empregados no tratamento de várias patologias inflamatórias. O mecanismo de ação dos AINEs envolve a redução da biossíntese de prostaglandinas (PG), por inibição direta da enzima prostaglandina endoperóxido sintase (PGHS)2.
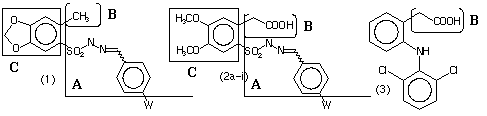
OBJETIVO: No âmbito de um programa de pesquisas que visa o planejamento, a síntese e a avaliação farmacológica de novos inibidores de enzimas da cascata do ácido araquidônico (AA), descrevemos anteriormente a síntese e atividade antiinflamatória dos derivados sulfonilidrazônicos (1), apresentando grupamentos ácidos na unidade benzilidênica (W), candidatos a agentes antiinflamatórios.3 Neste contexto, descrevemos nesta comunicação a síntese de uma nova família de derivados sulfonilidrazônicos fenilacéticos (2a-i), estruturalmente planejados explorando a hibridação molecular da série (1) com o diclofenaco (3), um AINE clássico, caracterizada pela introdução do grupamento farmacofórico carboxilato de (3) na subunidade B presente em (1), visando explorar interações coulômbicas com resíduos no sítio ativo da PGHS. Adicionalmente, o desenho estrutural da nova série de compostos (2a-i) respeitou a presença do grupamento farmacofórico sulfonilidrazona (A) e explorou o bioisosterismo dos resíduos aromáticos 3,4-metilenodioxifenila e 3,4-dimetoxifenila, a fim de identificar variações no reconhecimento molecular em função de suas contribuições como aceptores de ligação hidrogênio. A natureza dos substituintes W presentes na sub-unidade (A) dos derivados (2a-i), foi variada buscando-se investigar a participação de eventuais efeitos eletrônicos e lipofílicos desta sub-unidade estrutural na bioatividade destes compostos.3
(2a) W= H; (2b) W= Br; (2c) W= F; (2d) W= Cl; (2e) W= OCH3; (2f) W= CO2H; (2g) W= NO2; (2h) W= N(CH3)2; (2i) W= 3,5-ditertButil-4-OH
METODOLOGIA:
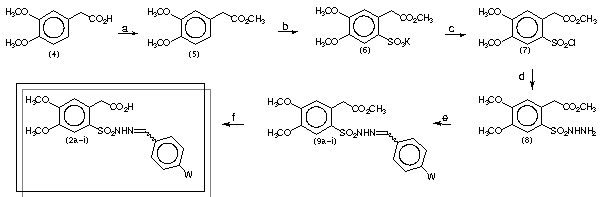
MeOH, H2SO4, 70oC, 4 h, 92%; b) 1- (CH3CO)2O, AcOEt, H2SO4, t.a., 3 h, 2- CH3CO2K, EtOH, t.a., 30min, 96%; c) SOCl2, DMF(cat.), 60oC, 3,5 h, 81%; d) N2H4.H2O, CHCl3, 0oC, 1,5 h, 75%; e) 4-W-C6H4CHO, EtOH, HCl, t.a., 1 h, 65-92%; f) MeOH/H2O, K2CO3, t.a., 24 h, 56-78%.
RESULTADOS: A síntese dos derivados sulfonilidrazônicos (2a-i) explorou como precursor o éster 3,4-dimetoxifenilacético (5) obtido em 92% de rendimento pela esterificação de Fisher do ácido correspondente (4). O derivado (5) foi funcionalizado regiosseletivamente na posição 6, através de substituição eletrofílica aromática, usando metodologia de clorossulfonação branda desenvolvida por Fraga e Barreiro4, que permitiu a obtenção do cloreto de sulfonila (7) em 77% de rendimento (2 etapas). Em seguida, a substituição nucleofílica quimiosseletiva de (7) com hidrazina hidrato, levou a obtenção da sulfonilidrazida (8) em 75% de rendimento. Condensação catalisada por ácido de (8) com benzaldeídos funcionalizados resultou na formação dos sulfonilhidrazonas correspondentes (2a-i), que foram transformadas nos compostos-alvo por hidrólise branda. Os derivados sulfonilidrazônicos (2a-i) obtidos apresentaram, majoritariamente, a configuração relativa E como evidenciado no experimento de nOedif, pela observação de nOe positivo pela irradiação do N-H hidrazônico no hidrogênio imínico.
CONCLUSÃO: A metodologia sintética empregada mostrou-se satisfatória, permitindo a obtenção dos derivados (2a-i) em rendimentos globais de 14% - 27%. A avaliação preliminar das propriedades antiinflamatórias no modelo de edema de pata de rato induzido por carragenina permitiu confirmar o perfil de atividade antecipado, com destaque para o derivado (2a) que apresentou 38,7% de inibição de formação do edema, sem apresentar ulcerogenicidade.
REFERÊNCIAS:
1- Emery, P. Drugs of Today (1999), 35, (4-5), 267; 2- Cryer, B. & Dubois, A. Prostaglandins & other Mediators (1998), 56, 341; 3- Lima, L. M. Pharm. Pharmacol. Commun. (1999), 5, 673; 4- Fraga, C. A. M. & Barreiro, E. J. J. Heterocyclic Chem. (1992), 29, 1667; 5- Fraga, C. A. M. Tese de Mestrado-IQ-UFRJ (1991); 6- Frölich, J. C. TiPS (1997), 18, 30; 7- Lima, L. M. Tese de Mestrado-IQ-UFRJ (1997); 8- Lima, P. C. Tese de Mestrado-IQ-UFRJ (1998).
APOIO FINANCEIRO: CNPq-PIBIC, CAPES, PRONEX