MODELAGEM MOLECULAR DE DERIVADOS N-ACILIDRAZÔNICOS COM AÇÃO ANTINOCICEPTIVA.
Fúlvia M. Santos(IC)1, Gabriela Barreiro(PG)1,2
Carlos R. Rodrigues(PQ)1 & Eliezer J. Barreiro(PQ)1
1Laboratório
de Avaliação e Síntese de Substâncias
Bioativas
(LASSBio, http://acd.ufrj.br/~pharma/lassbio),
Departamento de Fármacos, Faculdade de Farmácia,
Universidade Federal do Rio de Janeiro.
2Departamento de Química Orgânica, Instituto de Química,
Universidade Federal do Rio de Janeiro.
Palavras-chave : N-acilidrazonas, modelagem molecular, PM3.
Introdução
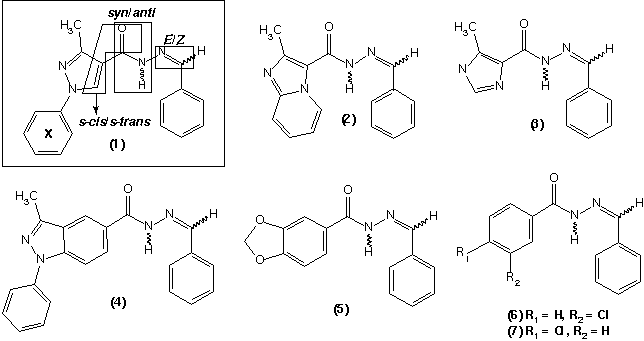
Dentro de um programa de pesquisas que visa o planejamento, síntese e avaliação farmacológica de compostos com ação antinociceptiva (ref), o derivado N-acilidrazônico (NAH) funcionalizado (1) foi sintetizado e apresentou potente atividade analgésica1. A partir de princípios de bioisosterismo, novos derivados NAH (2-7) foram planejados, sintetizados e bioavaliados, buscando-se estabelecer parâmetros que permitissem determinar os grupos farmacofóricos relevantes na interação com os nociceptores.
Objetivos
Neste trabalho visou-se estudar as contribuições estruturais e eletrônicos essenciais para atividade analgésica observada para as série de moléculas sintetizadas (2-7), utilizando-se métodos computacionais capazes de identificar as principais características estruturais relevantes na interação com o bioreceptor.
Tabela 1
|
Composto |
pKa |
LogP |
% de inibiçãoa |
|
(1) |
--- |
--- |
49,2 |
|
(2) |
5,56 |
1,23 |
5,2 |
|
(3) |
13,16 |
1,16 |
74,4 |
|
(4) |
5,54 |
3,19 |
52,0 |
|
(5) |
5,54 |
2,87 |
62,1 |
|
(6) e (7) |
5,56 |
3,37 |
7,0 |
aAtividade analgésica observada com a concentração de 100 mmol/kg, p.o. no teste de contorções abdominais induzidas por ácido acético 0,1N.
Metodologia Computacional
Todas os compostos (2-7) foram otimizados e, em seguida, analisados conformacionalmente utilizando-se as palavras-chave STEP=30 e POINT=12 para cada diedro de rotação livre, totalizando 12 confôrmeros para cada estrutura. Os cálculos foram realizados através do método semi-empírico PM3 inserido no
programa MOPAC 7.0. As estruturas de mínimo de energia foram re-otimizadas adotando-se as palavras-chave GNORM = 0,1, PRECISE e MMOK úteis na correção de ligações “peptídicas”. A superfície de energia potencial pôde ser calculada utilizando-se o programa Spartan. Os valores de pKa e logP dos compostos foram determinados através do programa PALLAS.
Para todos os compostos estudados foram identificadas oito conformações (s-cis/s-trans, syn/anti e E/Z, Figura), sendo a conformação syn predominante. Quanto à diastereoisomeria não foi possível detectar-se diferenças significativas na entalpia de formação das conformações s-cis/s-trans.
Os resultados farmacológicos indicaram (Tabela 1) que os compostos que não apresentam o anel aromático X, e.g. (3 e 5), evidenciaram atividade analgésica superior à observada para o composto (1)1,2,3. A ausência do grupamento fenila nos compostos mais ativos (3 e 5) pode estar potencializando a atividade, provavelmente pela menor restrição estérica que possuem. A baixa atividade observada para os compostos (6 e 7), pode ser interpretada pela ausência de grupamentos H-doadores/aceptores em contraste aos compostos mais ativos (3 e 5).
I. G. Ribeiro et al., Eur. J. Med. Chem., 33, 225-235 (1998).
C. Barja-Fidalgo et al., J. Pharm. Pharmacol., 51, 703-707 (1999).
L. F. C. C. Leite et al., Il Farmaco, 54, 747-757 (1999).
FAPERJ , CNPq, FUJB e PRONEX