SÍNTESE
DE NOVAS N-ACILIDRAZONAS IMIDAZÓLICAS PLANEJADAS COMO
NOVOS CANDIDATOS DE AGENTES ANALGÉSICOS.
Anna
Claudia Cunha1 (PG), Carlos Alberto Manssour Fraga1
(PQ), Maria Cecília Bastos Vieira de Souza2
(PQ), Vitor Francisco Ferreira2 (PQ) &
Eliezer Barreiro1
(PQ).
1-
Laboratório de Avaliação e Síntese de
Substâncias Bioativas (LASSBio), Faculdade de Farmácia-
UFRJ; 2- Instituto de Química- UFF.
Palavras-chave:
N-acilidrazonas, heterociclos, propriedades analgésicas
Introdução
Antiinflamatórios
não esteroidais (NSAI’s) são amplamente usados por
suas propriedades antiinflamatórias, analgésicas e
antipiréticas, além de antitrombóticas. O
mecanismo de ação dos NSAIS’s envolve a inibição
da biossíntese de prostaglandinas, a partir do araquidonato,
por ação direta ao nível da enzima
prostaglandina endoperóxido sintase (PGHS). Como os produtos
da PGHS expressada constitutivamente (prostaglandinas gástricas,
PGI2 e PGE2) são citoprotetores, em
especial para a mucosa gástrica, inúmeros pesquisadores
vêm procurando descobrir novos agentes terapêuticas que
possuam efeitos clínicos, mas que não apresentem
efeitos colaterais indesejáveis, principalmente os que levam
às ulceração gástrica e a nefrotoxidade.1
A
correta utilização do bioisosterismo, para modificação
molecular de compostos eleitos como protótipos, representa uma
estratégia atraente para o desenho estrutural de novos
candidatos eleitos a agentes terapeuticamente úteis, com
perfil farmacológico superior ao protótipo.
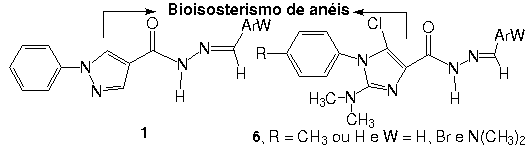
Desta
forma, novos derivados imidazólicos 6a-f foram
planejados racionalmente a partir de modificações
estruturais no protótipo pirazólico 1, descrito
como inibidor dual das enzimas PGHS e 5-LO. As modificações
estruturais introduzidas em 1, foram baseadas no
bioisosterismo clássico de aneis, onde o sistema pirazólico
de 1 foi trocado pelo sistema imidazólico de 6.
A função N-acilidrazona, importante para
atividade observada foi mantida visto sua natureza farmacofórica.



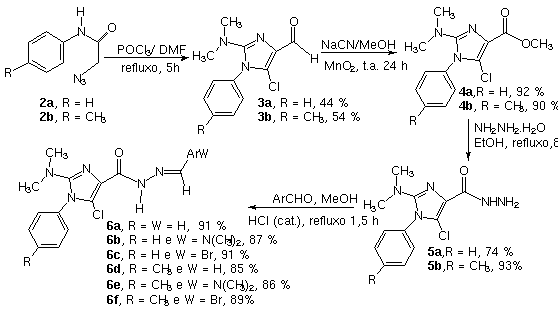
Esquema:
Síntese das N-acilidrazonas imidazólicas
6a-f
Resultados
e Discussão
A
introdução de substituintes em posição
para do grupamento fenila de 6a-f, foi planejada de
forma permitir avaliar as contribuições de propriedades
fisico-químicas e fatores eletrônicos nas atividades
analgésica e antiinflamatória.
A
rota sintética empregada na síntese das novas
hidrazonas imidazólicas 6a-f está ilustrada no
Esquema. O tratamento das azido-acetamidas funcionalizadas 2a-b
nas condições de Vielsmeier-Haack, forneceu os
imidazóis desejados 3a-b, que foram convertidos, em
altos rendimentos, nos ésteres metílicos 4a-c.
Subsequente interconversão do grupamento éster de 4a-b,
empregando-se métodos clássicos, permitiu a obtenção
dos intermediários-chaves 5a-f. Os compostos alvos 6a-f
foram preparados de forma diastereosseletiva, obtendo-se o
isômero (E) como único produto formado
nesta série de susbtâncias.
Conclusões
A
metodologia sintética empregada na síntese dos novos
derivados N-acilidrazônicos 6a-f permitiu sua obtenção
em bons rendimentos.
A
avaliação farmacológica preliminar dos
derivados 6a-c mostrou um perfil antiinflamatório
para 6a, inibindo o edema de pata de rato induzido por
carragenina em torno de 30%.
Bibliografia
1-
a) Barreiro, E. J.; Fraga, C. A.M.; Lages, A. S.; Romeiro, N. C.;
Quim. Nova, 1998, 21, 761; b) Janusz, J. M.; et. all;
J. Med. Chem.; 1998, 41, 1112
Agradecimentos:
FAPERJ