1
2
3
Estudos conformacionais de derivados do ácido 3-acilaminopicolínico e seus n-óxidos
Claudia Cristina Cardoso Bejana (PG), Ivoneide C. L.Barrosb (PG), João Bosco P. da Silvaa (PQ), Friedrich Wilhelm Joachim Demnitz (PQ) a, Hans-Ulrich Gremlichb (PQ).
aDepartamento de Química Fundamental, Universidade Federal de Pernambuco, Cidade Universitária, Recife-PE, Brasil; bDepartamento de Química, Universidade Federal de Amazonas, Manaus-AM, Brasil; cNovartis Pharma AG, Basel, Suíça.
palavras-chave: ácido 3-aminopicolínico, espectroscopia, análise conformacional.
A conformação de aromáticos orto-disubstituídos é fortemente influenciada pela interação entre os substituintes em questão. Entre os fatores que apresentam grande importância nestes casos são pontes de hidrogênio. Acetanilidas com receptores de pontes de hidrogênio na posição orto, assumem a conformação representada em 1 devido à ponte de hidrogênio existente entre N-H e X. Essa conformação obriga a carbonila da amida a assumir uma posição tal afim de desblindar o H-6 [1].
|
|
|
|
|
1 |
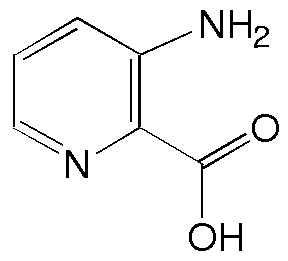
2 |
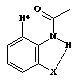
3 |
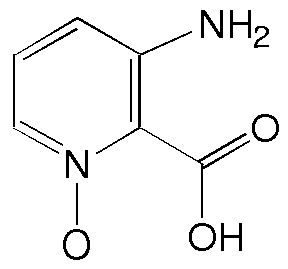
Recentemente sintetizamos derivados do ácido 3-aminopicolínico (2) e do seu N-óxido (3) como novos ligantes bidentados para complexação com íons lantanídeos. O grupo amino serve como ponto de introdução de substituintes apropriados para modificações nas propriedades físico-químicas e/ou eletrônicas de seus respectivos complexos. Para avaliar melhor os efeitos de substituintes na amina é fundamental um conhecimento conformacional dos derivados desta série.
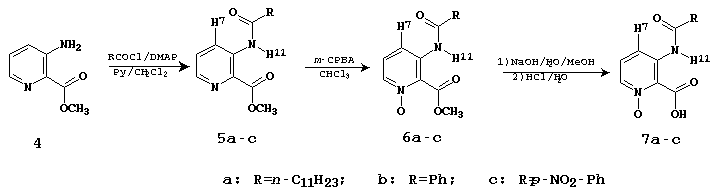
Para tanto, sintetizamos os ligantes 7a-c seguindo a rota sintética representada no Esq. 1. Assim como os produtos de interesse (7), os intermediários 5 e 6 foram então estudados por espectroscopia de 1H RMN e IV (em solução com CH2Cl2). Nestas estruturas, as interações entre o N-H11 e o substituinte carboxílico foram comparadas com as encontradas nas acetalinidas 1. A presença do grupo N-O nas estruturas 6 e 7, produzindo um sistema aromático 1,2,3-trisubstituído, resultou numa interação adicional influenciando a geometria molecular.
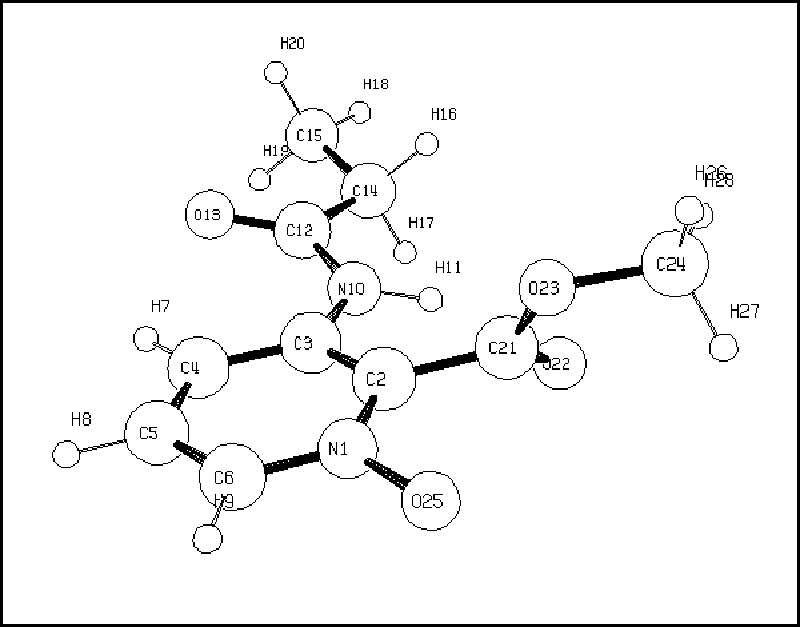
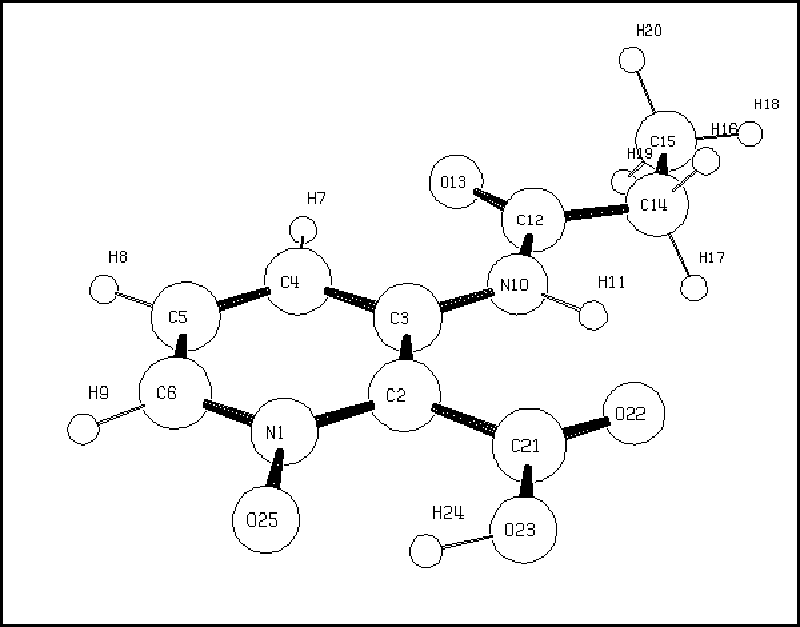
A Tab. 1 mostra os principais resultados que confirmam as conformações das moléculas 5-7, exemplificadas pelos derivados tipo a. Diante desses resultados podemos dizer que as estruturas 5 e 7 são completamente planares, indicando a presença de pontes de hidrogênio entre o N-H11 e o grupo C=O carboxílico. Este fenômeno é especialmente acentuado na estrutura 7 devido às pontes de hidrogênio existentes também entre o N-O e o O-H da carboxila. Enquanto isso a estrutura 6 apresenta uma forte repulsão eletrônica entre os grupos N-O e O-CH3 perturbando a planaridade da molécula e consequentemente diminuindo suas interações por pontes de hidrogênio.
As interpretações dos espectros foram substanciadas por resultados de cálculos ab initio RHF/6-31G, empregando como modelo as propanoilamidas respectivas. As estruturas mais representativas da modelagem molecular encontram-se nas Figs. 1 e 2. Como pode ser visto na Tab. 1, as estruturas otimizadas na modelagem molecular apresentam coerência com os resultados espectroscópicos de 1H RMN e IV.
Esquema 1:
|
Figura 1: modelo para 6a |
Figura 2: modelo para 7a |
Tabela 1:
|
|
1H RMN (ppm) d-H7 d-H11 |
IV (cm-1) nmax(N-H) |
Distância (Å) H11O22 H7O13 |
Ângulo Diedral N1C2C21O23 C4C3N10C12 |
|
4 |
7.03 5.75 |
|
|
|
|
5a |
9.15i 10.98ii |
3318v |
1.8565 2.1393 |
0.01o 0.04o |
|
6a |
8.36iii 8.83iv |
3401vi |
2.1442 2.2264 |
-59.86o 18.86o |
|
7a |
9.28 i 12.34ii |
3161v |
1.7881 2.0893 |
0.00o 0.01o |
i) alta desblindagem pelo C=O amida; ii) alta desblindagem pelo C=O carboxílico; iii) torção C4C3N10C12 Þ baixa desblindagem; iv) torção N1C2C21O23 Þ baixa desblindagem; v) ponte de hidrogênio forte; vi) ponte de hidrogênio fraca.
[1] Rae, I. D. Can. J. Chem., 1968, 46, 2589-2595.
CNPq e CAPES