1 Laboratório de Síntese, Departamento de Pesquisa e Desenvolvimento Tecnológico, Farmanguinhos, FIOCRUZ, R. Sizenando Nabuco 100, Manguinhos, Rio de Janeiro, CEP 21041-250, RJ; Brasil.
2 Farmacologia Aplicada, Farmanguinhos, FIOCRUZ, R. Sizenando Nabuco 100, 21041-250, RJ; Brazil.
Palavras-chave: tiadiazol, acilaminos, antineoplásico,
Diante da necessidade de novas drogas eficazes para a quimioterapia antineoplásica. Este trabalho tem como objetivo a síntese de derivados 1,3,4-tiadiazólicos procurando manter os níveis de atividade e reduzir a toxidez. Os produtos obtidos tiveram a sua atividade avaliada in vitro.
Desta forma a partir do 2-amino-5-mercapto-1,3,4-tiadiazol utilizamos como estratégia a síntese de derivados acilaminos utilizando como agentes acilantes os anidridos acético, glutárico e succínico, sendo sintetizados e estudados também na forma de sulfetos e sulfonas, ver Esquema1.
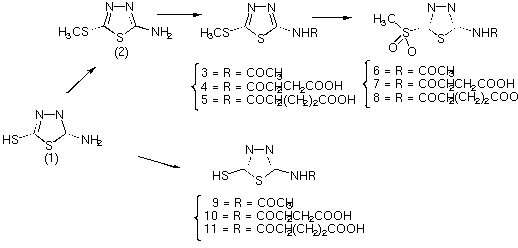
Os produtos obtidos foram caracterizados através de espectros de IV, 1H-RMN e 13C-RMN e enviados para a farmacologia a fim de terem sua atividade antineoplásica in vitro avaliada. Os resultados obtidos são apresentados na Tabela1.
|
Produto |
Cito |
J774 |
SP20 |
NEURO-2A |
MK2 |
CARC |
BWP36535OK36,1181,4564,8146,28-88,62NEG6OK49,4582,53NEGICCNEG82,36NEG8OK62,7885,5156,9542,93NEG81,75NEGTabela 1: Cito–citotoxicidade, NEG–negativo, ICC–indução de crescimento celular. Linhagens murinas: J774–macrófago, SP20–mielona, NEURO-2A–neuroblastoma, CARC (carcinoma de erlich)–sarcoma induzido por metil colantreno, BW–timona, P3653–plasmocitona. MK2–célula epitelial de rim de macaco.
|
|||||||
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|||||||
O teste utilizado foi o de incorporação de sal de tetrazolium (MTT) às mitocôndrias das células, em que 105 células/poço foram incubadas com as diferentes drogas na concentração de 2,5 mg/mL, sendo o ensaio feito em triplicata. Após 48 horas as células foram submetidas à incubação com MTT e o resultado expresso em percentual de atividade em relação ao controle negativo (células incubadas com meio RPMI). Sendo considerandos significativos os resultados acima de 50%. O teste de citotoxicidade realizado com hemácias de carneiro, demonstrou que nenhum dos análogos foi tóxico. Com esses resultados preliminares (Tabela 1), podemos concluir que os derivados 5, 6 e 8, que foram testados até agora, apresentaram atividades antineoplásicas elevadas frente as células SP20 e BW.
Korolkovas, A., Burckhalter, J.H., Química Farmacêutica, 1982, 618-648.
Lu, S.M., Chen, R.Y., Synthetic Communications, 1999, 29, 3443-3450.
Ciotti, M.M., Humphreys, S.R. et al, Cancer Research, 1960, 20, 1195-1201.
Oleson, J.J., Sloboda, A. et al, J. Am. Chem. Soc., 1955, 77,6713-6714.