Síntese de Novos DERIVADOS 1,3,4-TIADIAZÓLICOS PLANEJADOS COMO lEISHMANICIDAS, ANÁLOGOS À COMBRESTATINAS nATURAIS
Aline Guerra Manssour Fraga1 (PQ), Victor Brasil3(PG), Marcelo Bozza3 (PQ), Edson Ferreira da Silva1 (PQ), Carlos Alberto Manssour Fraga2 (PQ)
1 Laboratório de Síntese, Departamento de Pesquisa e Desenvolvimento Tecnológico, Farmanguinhos, FIOCRUZ
2 Departamento de Fármacos, Faculdade de Farmácia, UFRJ
3 Farmacologia Aplicada, Farmanguinhos, FIOCRUZ, R. Sizenando Nabuco 100, 21041-250, RJ; Brazil.
palavras-chave: leishmaniose, derivados 1,3,4-tiadiazólicos, combrestatinas
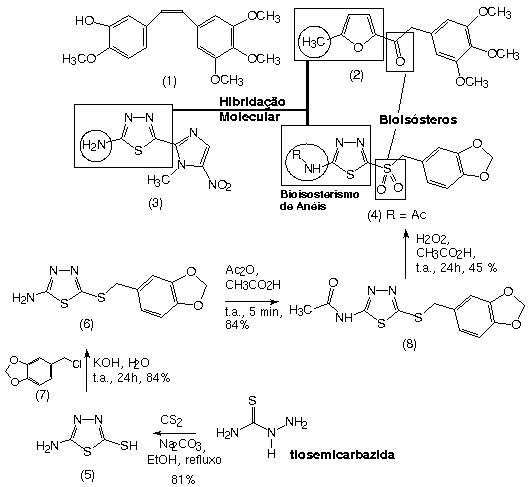
A leishmaniose humana afeta 12 milhões de pessoas em todo o mundo, apresentando uma incidência anual de novos casos de ca. de 2 milhões de pessoas. Esta infecção é causada por protozoários do genero Leishmania que são transmitidos por insetos hematófagos conhecidos como flebotomíneos. A leishmaniose apresenta-se em diferentes formas clínicas (visceral, cutânea ou mucocutânea1), dependentes tanto de espécie de Leishmania quanto da imunidade do hospedeiro. Atualmente a leishmaniose tem sido tratada com drogas antimoniais pentavalentes ou antibióticos como a anfotericina B, fármacos de elevada toxicidade que induzem efeitos colaterais indesejáveis, limitando seu emprego clínico2. As combrestatinas, e.g. combrestatina A4 (1), são produtos naturais de grande interesse terapêutico devido às suas propriedades anti-mitóticas2, úteis no tratamento de alguns tipos de câncer. Recentemente, del Rey e col.3 descreveram a atividade leishimanicida de combrestatinas sintéticas, dentre as quais merece destaque o derivado furânico (2) que apresentou IC50 = 0,48 mg/mL contra L. brasiliensis. Adicionalmente, merece destaque na terapia anti-protozoário o megazol4 (3) derivado 1,3,4-tiadiazólico que apresenta expressiva atividade tripanomicida. Neste contexto, este trabalho tem como objetivo a síntese e a avaliação das propriedades leishmanicidas de um novo derivado 1,3,4-tiadiazólico (4), planejado pela hibridação molecular da combrestatina sintética (2) com megazol (3). O derivado (4) foi desenhado explorando os princípios de bioisosterismo clássico de anéis, além da substituição bioisostérica da carbonila presente em (2) pelo grupamento isóstero sulfona de (4). Adicionalmente, visando reduzir o caráter toxicofórico do grupamento amino heteroaromático presente em (3)5, o derivado acetilado correspondente, i.e. (4), foi identificado como candidato à leishmanicida com baixo potencial de toxicidade.
A síntese dos compostos-alvo utilizou como matéria-prima o derivado 2-mercapto-5-amino-1,3,4-tiadiazola (5) obtido em 81% de rendimento, segundo metodologia descrita por Petrow6, explorando a condensação do dissulfeto de carbono com a tiosemicarbazida. Em seguida, (5) foi quimiosseletivamente convertido no tio-éter (6), em 84% rendimento, pelo tratamento com o cloreto benzílico7 (7), usando KOH aquoso como base7. Proteção de grupamento amino de (6) pelo tratamento com anidrido acético à temperatura ambiente7 levou a obtenção do derivado acetilado (8) em 84% rendimento. Finalmente, o sulfeto (7) foi oxidado à sulfona correspondente (4), em 45% de rendimento, pelo tratamento com H2O2 e ácido acético à temperatura ambiente8.
O resultado da avaliação das propriedades leishmanicidas do derivado (4), bem como dos precursores sintéticos (6) e (8), foram avaliados quanto ao efeito leishmanicida em ensaios in vitro utilizando-se promastigotas de L. major nas concentrações de 100, 10, 1 e 0,1mg/ml. Estes compostos não apresentaram atividade leishmanicida significante no tempo de 24h de tratamento.
Referências:
1- Genaro, O. Leishmaniose Tegumentar Americana, in “Parasitologia Humana”. Neves, D.P., Ed., Atheneu, São Paulo, 1995, pp. 41-60.; 2- http://www.who.int/emc/diseases/leish ; 3- del Rey, B. et al. (1999) Bioorg. Med. Chem. Lett. 9 2705-2710.; 4- Enanga, B. et al. (1999) Arzneim Forsch 49 (I), 441-447; 5- Oleson, J.J et al. (1955) J. Am. Chem.Soc.77, 6713-6714; 6- Petrow, V. et al. (1958) J. Chem. Soc , 1508-1513; 7- Young, R.W. et al. J.Am.Chem.Soc. 78(1956), 4649-4654.; 8- Vampa, G. etal. (1995) J. Heterocyclic Chem. 32, 227-234.
CNPq/FIOCRUZ/FAPERJ