Valderi Luiz Dressler(PQ), Érico Marlon de Moraes Flores(PQ), José Neri Paniz(PQ) Ayrton Figueiredo Martins(PQ), Daniel Bueno Boeira(IC) e Juliano Barin(IC)
Departamento de Química - Universidade Federal de Santa Maria - RS
Palavras-Chave: CVAAS, peixe, mercúrio.
O mercúrio (Hg) é reconhecidamente um elemento altamente tóxico e presente num grande número de alimentos, sendo, então, a ingestão destes, uma das principais fontes de intoxicação. Entre os alimentos consumidos pelo homem, os peixes, são, talvez, os produtos de origem animal que mais contém Hg. Devido à considerável toxicidade do elemento, foi estipulado que a ingestão média semanal de Hg (total) seja de, no máximo, 300 µg. Esta quantidade pode facilmente ser atingida com uma dieta rica em peixes, principalmente, se estes são provenientes de locais contaminados.1 Para a determinação de Hg, a técnica de absorção atômica (AAS) com geração de vapor frio (CV) é uma das mais recomendadas, principalmente por ser sensível, seletiva, simples e de baixo custo. Nesta técnica, mercúrio elementar (sob a forma de vapor) é gerado a partir da redução de seus íons em solução, através do emprego de cloreto estanoso (SnCl2) ou borohidreto de sódio (NaBH4), em meio ácido. Este processo pode ser executado através do uso de geradores em batelada, ou sistemas de fluxo contínuo ou sistemas de injeção em fluxo (FI).2
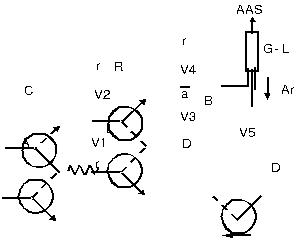
Neste trabalho é proposto estudo relacionado ao desenvolvimento de metodologia analítica para a determinação de Hg em amostras de peixe, empregando-se um sistema FI (Fig. 1) acoplado a técnica CVAAS.
O sistema basicamente consiste de um espectrômetro de absorção atômica, 5 válvulas solenóides de três vias, uma bomba peristáltica de 8 canais (equipada com tubos de Tygon de diversos diâmetros), tubos de Teflon (d.i = 0,8 mm) e um separador gás-líquido de polietileno (L= 10 cm e d.i. = 1 cm). ). O funcionamento do sistema FI dá-se da seguinte maneira: inicialmente a válvula V1 é acionada (07 s) de forma que a alça de amostragem L seja preenchida com amostra ou solução analítica (A), posteriormente são acionadas as válvulas V2, V3 e V4 (08 a 25 s) permitindo que a alíquota da amostra seja misturada com o redutor na confluência “a” e, finalmente, a válvula V5 é acionada (25 a 50 s) para remover a solução residual do separador gás-líquido.
Parâmetros relacionados com o sistema FI, tais como, vazão da solução do redutor (0,5 a 2,0 mL), da solução carregadora (2 a 8 mL) e do gás de arraste (0,2 a 1,0 mL), concentração do redutor (0,25 a 5% m/v) e do ácido (HNO3 de 0 a 30% v/v) usado como diluente das soluções analíticas, comprimento do reator (B) e volume da amostra (L), foram testados de modo a definir as melhores condições de desempenho do sistema. Testes também foram feitos em relação ao uso de água, HCl e HNO3, como solução carregadora da amostra.
As melhores respostas do sistema foram obtidas com as seguintes vazões: solução carregadora em torno de 5,0 mL min-1, solução redutora a 0,8 mL min-1 e gás de arraste em 0,3 L min-1, sendo esta última bastante crítica para a repetibilidade das medidas. Nestas condições verificou-se que o sinal analítico não sofre grande influência da variação da concentração do redutor na faixa de 2,0 a 4,0% m/v, bem como, da concentração de HNO3 na faixa de 0,5 a 30% v/v (este usado no preparo das soluções de calibração). Mesmo obtendo-se sinais analíticos semelhantes entre o HCl e HNO3 usado como solução carregadora da amostra, optou-se pelo uso do HNO3 0,2% v/v. Entretanto, flutuações elevadas do sinal foram verificadas quando do uso de água como carregador da amostra, possivelmente devido a variações muito grandes na acidez das soluções.
Nas condições empregadas, o desvio padrão relativo do método (RSD) para 10 leituras consecutivas da amostra foi de 5%, limite de detecção (LD) de 0,10 µg g-1, (calculado a partir de 3 s/S, sendo s o desvio padrão de 10 leituras do branco da amostra e S a inclinação da curva de calibração) e m0 de 58 pg de Hg. A exatidão do método foi verificado através do uso de material de referência certificado (Fish Tissue-IAEA MA-B-3/TM), obtendo-se resultados concordantes com os certificados. Também, tendo-se em vista que pode haver diferença de composição do material de referência e das amostras reais analisadas, fez-se testes de recuperação do analito. Nestes testes, as recuperações foram todas próximas de 100%. Três amostras reais de peixe foram analisadas pelo método proposto: peixe castanha (in natura) cuja concentração de Hg medida foi 0,046 ± 0,005 µg g-1; atum beneficiado (enlatado) com concentração de 0,184 ± 0,017 µg g-1 e sardinha beneficiada (enlatada). Nesta amostra, a concentração medida foi menor do que o LD do método. Assim, conclui-se que o método proposto é adequado para a determinação de Hg em amostras de peixe, apresentando boa exatidão e repetibilidade.
Figura 1. Diagrama do sistema FI. A: amostra; C: carregador (HNO3 0,2%); R: redutor (NaBH4); L: alça de amostragem; B: reator (15 cm); D: descarte: r: reciclo de solução; a: confluência; V: válvulas solenóides; Ar: argônio: G-L: separador gás-líquido; AAS: espectrômetro de absorção atômica.
Tsalev, D. L.and Zaprianov, Z. K. Atomic Absorption Spectrometry in Occupational and Environmental Health Practice. Volume I, II e III, CRC, Boca Raton, Florida, USA, 1985.
Welz, B. Atomic Absorption Spectrometry. 3nd ed., VCH, Veinheim-Germany, 1985, 506 p.
FAPERGS, CNPq