UTILIZAÇÃO DO ALGORITMO GENÉTICO NA DETERMINAÇÃO DO ALINHAMENTO DE COMBILEXINAS DERIVADAS DA ACRIDINA
Andrei Leitão (IC), Carlos Alberto Montanari (PQ)
Núcleo de Estudos em Química Medicinal, Departamento de Química, Universidade Federal de Minas Gerais
palavras-chave: algoritmo genético, alinhamento, combilexinas
Novas metodologias são continuamente desenvolvidas e aperfeiçoadas, num esforço para o avanço científico na fundamentação e consolidação da utilização de estudos teóricos aliados às necessidades propostas em diferentes áreas do conhecimento.
O algoritmo genético tem demonstrado, juntamente com outras técnicas, seu valor real referente à sua utilização em alinhamentos de moléculas. Esta técnica utiliza conhecimentos acumulados da reprodução dos seres vivos e seleção de espécies para a resolução de diversas questões, incluindo o alinhamento de moléculas.
O alinhamento de moléculas é um passo fundamental para o desenvolvimento de uma metodologia confiável para a utilização em QSAR clássico e QSAR-3D e tornou-se necessário para as moléculas com atividade anticancerígena, como a acridina e seus derivados, as combilexinas.
Estas moléculas, submetidas à modelagem molecular no programa MacroModel, serviram como ponto de partida para o alinhamento no programa GASP, dentro do pacote de programas Sybyl.
Foi definida uma molécula base, sendo esta molécula a mais potente do conjunto utilizado. A molécula base foi mantida rígida durante o cálculo, enquanto as outras se ajustaram a esta molécula.
Alguns parâmetros especificados como fundamentais pelo programa, como a energia de van der Waals, a ionização na molécula, a capacidade de doar ou receber elétrons, a presença de par de elétrons não ligante, o volume de superfície, promovem a seleção das moléculas através do algoritmo genético. Este algoritmo genético tem, por sua vez, as propriedades de permutação ("crossover"), mutação e a própria seleção, onde estes dados são armazenados na forma de cromossomas moleculares, atuando livremente durante todo o processo de cálculo.
Um conjunto de medidas foi tomado visando a obtenção de um equilíbrio entre a confiabilidade do cálculo e o tempo computacional gasto. Entre as ações que podem ser utilizadas com esta finalidade pode-se enumerar o aumento da importância da seleção, redução da mutação e permutação, aumento do mínimo a ser considerado para a energia de van der Waals, dentre outros parâmetros que podem ser facilmente alterados.
O cálculo efetuado procurou aliar a necessidade do uso das regras de ionização que o programa trabalha, pois existem moléculas ionizadas, com a presença de uma diversidade de conformações bem grande. Estes dois fatores podem gerar problemas quanto ao tempo computacional despendido, não havendo a convergência do cálculo se não houver uma correção.
Diversos ajustes foram efetuados na tentativa de uma melhor solução, pois o programa não possui regras estritamente definidas para o cálculo, apenas algumas ações são delineadas como pontos referenciais em relação ao tempo e à qualidade do cálculo.
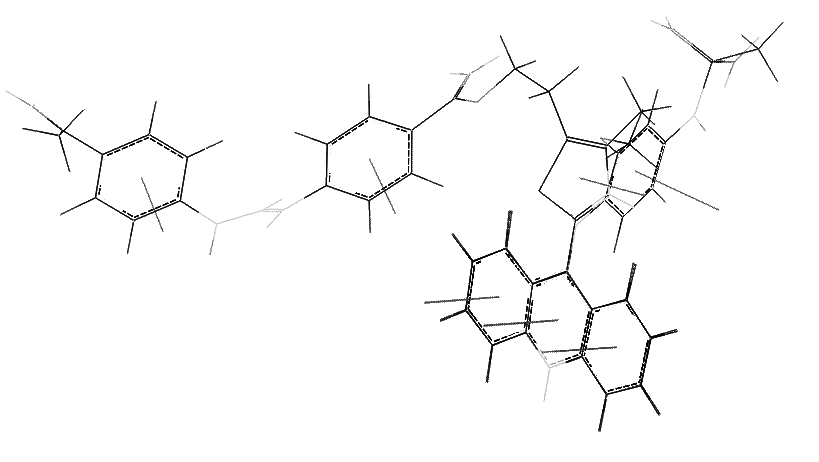
Os alinhamentos foram efetuados considerando-se pequenos conjuntos de moléculas (três ou quatro) para as que continham mais graus de liberdade e acima disto para as que continham menos graus. Desta forma as 19 moléculas foram divididas em 7 conjuntos, contendo em todos eles a molécula base, a m-amsacrina (como representado na figura 1).
Figura 1 - Superposição de algumas estruturas alinhadas no programa GASP, usando-se o algoritmo genético
O alinhamento feito permite a utilização destas moléculas como ponto de partida para o uso em técnicas de QSAR, fundamentais no estudo de novos fármacos na química medicinal.
Referências Bibliográficas:
1. Jenkins, T. C.; McConnaughie, A. W.; Journal of Medicinal Chemistry, (1995), 38, 3488-3501
CNPq/FAPEMIG/FINEP/PRPq