ESTUDOS VISANDO A SÍNTESE ENANTIOSSELETIVA DA (-)-ARCTIGENINA
André Ricardo Gomes Ferreira (PG),1 Ayres Guimarães Dias (PQ),2 Paulo Roberto Ribeiro Costa (PQ).1
Laboratório de Síntese Assimétrica (LASA), Núcleo de Pesquisa de Produtos Naturais, Universidade Federal do Rio de Janeiro - Brasil.
Departamento de Química Orgânica, Instituto de Química, Universidade do Estado do Rio de Janeiro - Brasil.
palavras-chave: lignana, síntese enantiosseletiva e adição conjugada.
Introdução: As lignanas apresentam uma vasta ocorrência na natureza e um variado perfil de atividades biológicas tais como, antitumoral, antiviral, anti-PAF, anti-hepatotóxica e diurética.1 Entre as usadas em clínica destacamos a podofilotoxina, que vem sendo empregada em uso tópico no tratamento de câncer de pele, o etoposídeo e o tenoposídeo que são derivados semi-sintéticos da podofilotoxina usados como agentes antineoplásicos em tumores testiculares, câncer pulmonar de células pequenas e usado como um dos componentes de tratamentos antineoplásicos pluriterapêuticos no combate a leucemia linfoblástica aguda da infância. Por outro lado a (-)-Arctigenina, isolada de espécies Forsythia, apresenta ação citotóxica in vitro contra células HepG2, atividade anti leucêmica em células mielóides de ratos, inibição do crescimento de células HL-60 com IC50 menor que 100ng/mL, quase a dose de inibição dos agentes anticâncer utilizados clinicamente. Esta lignana inibe também a replicação do HIV-1 em células humanas infectadas.
Objetivo: Nossos esforços estão concentrados na síntese enantiosseletiva de dibenzilbutirolactonas, em particular a (-)-Arctigenina (7). Doyle e colaboradores sintetizaram recentemente o seu enantiômero, a (+)-Arctigenina.2
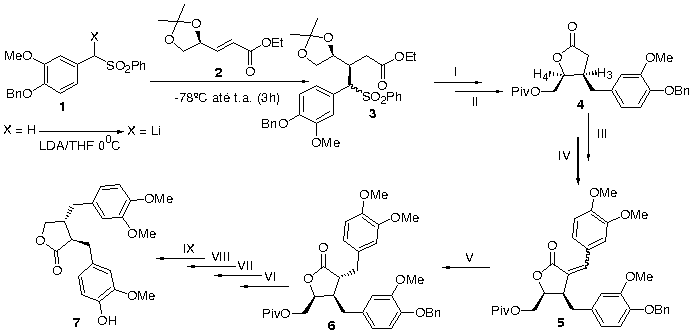
Método: Estudos metodológicos foram desenvolvidos por nosso grupo em adições conjugadas de nucleófilos a enoatos quirais g-oxigenados derivados do D-(+)-Manitol.3 O uso de sulfonas como carreadoras de unidades alílicas, prenílicas e benzílicas foi investigado, conduzindo a produtos de adição tanto sin quanto anti (ed>90%).4 Para a obtenção de 7, utilizaremos a sulfona 1, derivada da Vanilina benzilada, como carreadora da unidade C6-C1. A adição conjugada ao enoato 2, deverá levar a formação do aduto sin 3, com a configuração idêntica a do carbono 2 da lignana alvo.
Resultados: Os adutos obtidos pela reação de 2 com sulfonas preparadas a partir do piperonal e do 3,4-dimetoxi-benzaldeído, (-78ºC —> t.a., 2h), foram obtidos de forma sin com ed>95:5 e total controle no centro metino sulfônico. A sulfona 1 feita reagir com 2, nas mesmas condições, levou a mistura de adutos sin:anti (1:1). O aumento do tempo de reação ocasionou um aumento de produtos de degradação sem influenciar na estereosseletividade. Alternativamente, a sulfona 1 foi então desprotonada com LDA -780C e o carbânion feito reagir com o enoato 2, permanecendo a temperatura ambiente por 3h. O aduto de Michael 3 foi obtido em 60% de rendimento e apresentou-se como uma mistura de diastereoisômeros (sin:anti = 85:15), com total controle no centro metino sulfônico em cada aduto.
Na(Hg)6%/MeOH, II- a) HCl 20%/MeOH; b) Et3N, (CH3)3COCl, III- LDA/THF, 3,4-dimetoxi-benzaldeído, Et3N, H3CCO2COCH3, IV- DBU/Benz.-ref., V- NiCl2.6H2O-NaBH4, VI- LiAlH4/THF, VII- NaIO4/THF, VIII- PCC, IX- H2/Pd
O aduto 3 foi dessulfurizado (75%) seguido de tratamento em meio ácido, e proteção da hidroxila com cloreto de pivaloíla (75%, duas etapas), originando a lactona 4, majoritária. A relação cis entre os substituintes do anel lactônico foi caracterizada pela constante de acoplamento entre H3 e H4 (J = 7,12Hz), confirmando assim que o aduto majoritário 3 é proveniente de uma adição sin. A reação de condensação aldólica com a OMe-Vanilina, seguida de acetilação in situ levou a uma mistura de diastereoisômeros em 75%, que após tratamento com DBU em benzeno refluxo, conduziu a formação de um único produto 5 com uma ligação dupla exocíclica, em 83% de rendimento. Posterior redução com NaBH4 e NiCl2.6H2O proporcionou a formação da lactona 6 trisubstituída nas posições 2, 3 e 4 (cis - trans). As etapas subseqüentes para a síntese de 7 estão em andamento.
Conclusões: O método desenvolvido foi altamente estereosseletivo e conduziu a intermediários avançados na síntese de 7 a partir de matérias-primas abundantes.
Bibliografia:
Ward RS, Nat. Prod. Rep. 1999, 16, 75.
Doyle MP, Bode JW, Protopova MN, Zhou QL, J. Org Chem. 1996, 61, 9146.
Costa JS, Dias AG, Anholeto Al, Monteiro MD, Patrocínio VL, Costa PRR, J. Org Chem. 1997, 62, 4002.
Ferreira ARG, Dias AG, Pinto AC, Costa PRR, Miguez E, da Silva AJR, Tetrahedron Lett. 1998, 39, 5305.
CNPq, PRONEX, FUJB, UFRJ, UERJ.