RMN NA DETERMINAÇÃO DA CONFIGURAÇÃO ABSOLUTA DE CENTROS QUIRAIS EM BUTIROLACTONAS DERIVADAS DO D-(+)-MANITOL.
André Ricardo Gomes Ferreira (PG),1 Ayres Guimarães Dias (PQ),2 Eduardo Miguez (PQ),3 Antônio Jorge Ribeiro da Silva (PQ),3 Paulo Roberto Ribeiro Costa (PQ).1
Laboratório de Síntese Assimétrica (LASA), Núcleo de Pesquisa de Produtos Naturais, Universidade Federal do Rio de Janeiro - Brasil.
Departamento de Química Orgânica, Instituto de Química, Universidade do Estado do Rio de Janeiro - Brasil.
Central Analítica-Núcleo de Pesquisa de Produtos Naturais, Universidade Federal do Rio de Janeiro – Brasil.
palavras-chave: rmn, butirolactonas e configuração absoluta.
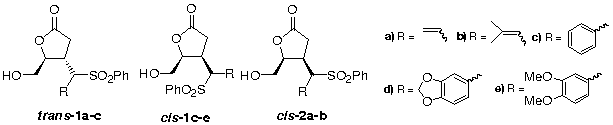
Introdução: Reações de adição conjugada estereosseletiva de nucleófilos a aceptores derivados do D-(+)-Manitol e do ácido L-Tartárico vêm sendo estudadas em nosso laboratório,1-3 dentro do contexto de um programa visando a síntese enantiosseletiva de produtos bioativos. A adição de derivados da fenilsulfona a enoatos derivados do Manitol produz adutos onde dois novos centros quirais são gerados.2 Estes produtos podem ser transformados nas respectivas lactonas, onde a configuração dos novos centros quirais pode ser determinada por medida diferencial de efeito Overhauser nuclear e pelas constantes de acoplamento H-H. As lactonas trans-1a,b e cis-2a-b tiveram as suas estruturas elucidadas desta forma.3
Objetivo: Determinação da configuração dos centros quirais em C3 e C1’, em lactonas substituídas por diferentes grupos benzila (cis-1c-e).
Método: As lactonas cis-1c-e foram preparadas a partir dos correspondentes adutos oriundos da adição conjugada da benzilfenilsulfona e derivados substituídos por grupos benzílicos oxigenados. As medidas diferenciais do efeito Overhauser nuclear para 1H foram feitas a 200MHz. O pulso de 90 para 1H foi de 24s. Nas medidas de eOn, o tempo de irradiação por sinal usado foi de 100 ms tipicamente e um tempo de espera de 4s foi utilizado antes de cada nova repetição da seqüência. As amostras foram preparadas em clorofórmio deuterado a 1-1,5% e a 300C e desgaseificadas por passagem de uma corrente de N2 através das soluções. Os valores de T1 foram medidos pelo uso da seqüência “Inversion-Recovery” para os carbonos a 100 MHz em solução a 2,5% preparada com CDCl3.
Resultados: Buscando subsídeos que pudessem corroborar os assinalamentos prévios das configurações das lactonas trans-1a,b e cis-2a,b, foram medidos os valores para os tempos de relaxação longitudinal dos carbonos do acetato da lactona cis-2a. Os resultados obtidos (T1 = 0,8s; C3, C4 e C1’) indicaram um predomínio do mecanismo de relaxação dipolar (tempos curtos medidos e relação entre (T1(CH) = 2xT1(CH2)) o que indicaria reorientação isotrópica em solução para esta molécula, e reduzida movimentação dos segmentos substituintes em C-3, C-4 e C-1’, validando o uso do eOn como ferramenta para estas determinações estruturais. Nas novas moléculas sintetizadas (trans-1c e cis-1-c-e), um anel aromático está ligado diretamente ao centro C1’ e outro tem o grupo sulfona como espaçador. A distinção entre os hidrogênios dos dois anéis aromáticos, fundamental para assinalamento dos centros quirais em C1’, baseam-se em estudos de eOn da fenilbenzilsulfona. A irradiação dos hidrogênios benzílicos apresentou eOn de 4,09% para os hidrogênios aromáticos do anel sem sulfona. Considerando as observações feitas para a lactona cis-1a como válidas para a atual série de moléculas, foi possível determinar a configuração dos carbonos C-3 e C-1’ nas lactonas trans-1c e cis-1-c-e com base nos acoplamentos vicinais JH-3,H-4 (7,67 a 7,74Hz) e JH-3,H-1’, (10,1 a 11,6Hz) e medidas de eOn envolvendo H-2, H-3, H-4, H-5, H-1’ e os orto H na fenilsulfona.

Conclusões: O uso do conjuntos de dados gerados pelas medidas de efeito Overhauser nuclear diferencial e das constantes de acoplamento JH-3,H-4 (7,67 a 7,74Hz) e JH-3,H-1’, (10,1 a 11,6Hz), permitiu a proposição das configurações nos carbonos C3 e C1’ das lactonas 1-c-e .
Bibliografia:
Costa JS, Dias AG, Anholeto Al, Monteiro MD, Patrocínio VL, Costa PRR, J. Org Chem., 1997, 62, 4002.
Dias AG, Ferreira ARG, Pinto AC, Costa PRR, 8th Brazilian Meeting on Organic Synthesis,1998, PS-041, 88
Ferreira ARG, Dias AG, Pinto AC, Costa PRR, Miguez E, da Silva AJR, Tetrahedron Lett., 1998, 39, 5305.
CNPq, PRONEX, FUJB, UERJ.