SIMULAÇÃO
DINÂMICA MOLECULAR DE ACETATO DE LÍTIO (LiAc)
L. S. Barreto1,2,3, S. R. Ivins2, K. A. Mort
2,
R.A. Jackson2, O. L. Alves1
1Laboratório de Química do Estado Sólido
– Instituto de Química – UNICAMP
C.P. 6145,
CEP: 13081-970, Campinas, SP, Brasil
led@iqm.unicamp.br
2Chemistry
Department – Keele University – Staffordshire – ST5
5BG (UK)
3Departamento
de Química – UFS – CEP: 49100-000 São
Cristovão – SE
Palavras-chave:
dinâmica molecular, acetato de lítio, transição
de fase
A
simulação dinâmica molecular (DM) tem sido uma
ferramenta útil para o estudo de líquidos orgânicos.
Diversos trabalhos foram desenvolvidos para moléculas pequenas
como haloalcanos, butanos, entre outros, reproduzindo parâmetros
experimentais como densidade, entalpia de vaporização e
estrutura intermolecular1. Essa técnica tem sido
também, aplicada ao estudo de fases cristalinas, cristais
líquidos, e vidros para os quais podem ser obtidos parâmetros
de ordem, constantes elásticas e propriedades de transporte2.
Estudos
de capacidade calorífica, Infravermelho e Ressonância
Magnética Nuclear de 23Na de vidros carboxilatos
de metais alcalinos indicaram que as estruturas das fases cristalinas
e vítreas não apresentam diferenças marcantes na
ordem a curta distância3,4. As transições
de fase para formação da fase vítrea são
complicadas pela possibilidade de formação de fases de
cristal líquido.
A técnica
de DM se apresenta como uma ferramenta adicional ao estudo dos vidros
de carboxilatos metálicos para obter informações
estruturais que permitam uma interpretação mais acurada
dos resultados experimentais.
No
presente trabalho foi utilizado o programa de simulação
dinâmica molecular DLPOLY5 para gerar um modelo que
descreva a estrutura do acetato de lítio cristalino, líquido
e sua fase vítrea.
Resultados
de difração de raios-X sugerem que o LiAc pertence ao
grupo espacial P com parâmetros de cela: a= 9,29 Å, b= 12,33 Å and
c= 6,80Å, a= 101,0 o,
b= 100,19 o, g=
105,5o. A densidade de 1,227 g/cm3, com 8
moléculas por cela unitária e volume de 716,1 A3.
São sugeridos quatro possíveis conjuntos de coordenadas
fracionárias atômicas para carbono e oxygênio,
negligenciando-se as posições do lítio e dos
hidrogênios6.
com parâmetros de cela: a= 9,29 Å, b= 12,33 Å and
c= 6,80Å, a= 101,0 o,
b= 100,19 o, g=
105,5o. A densidade de 1,227 g/cm3, com 8
moléculas por cela unitária e volume de 716,1 A3.
São sugeridos quatro possíveis conjuntos de coordenadas
fracionárias atômicas para carbono e oxygênio,
negligenciando-se as posições do lítio e dos
hidrogênios6.
A configuração
de partida usada foi a estrutura cristalográfica do acetato de
sódio anidro pela substituição dos íons
sódio por lítio. Os potenciais utilizados foram
transferidos de estudos de minimização de energia e DM
para fases cristalinas do acetato de lítio hidratado e acetato
de sódio7. As condições de equilíbrio
foram estabelecidas dentro de uma variação
estatisticamente aceitável para os valores de energia,
temperatura e pressão durante a simulação em
NPT-constante. O número de átomos foi de 1728. O time
step foi de 3x10-3 ps.
As variações
nas propriedades termodinâmicas e estruturais mostram uma
transição de fase de primeira ordem. A inflexão
na curva de entalpia indica absorção de calor latente,
Fig. 1. A função de distribuição radial,
Fig. 2, mostra que com o aquecimento as moléculas se difundem,
preservando ordem a curta distância porém, não
apresentam ordenamento a media e longa distância (MRO e LOR).
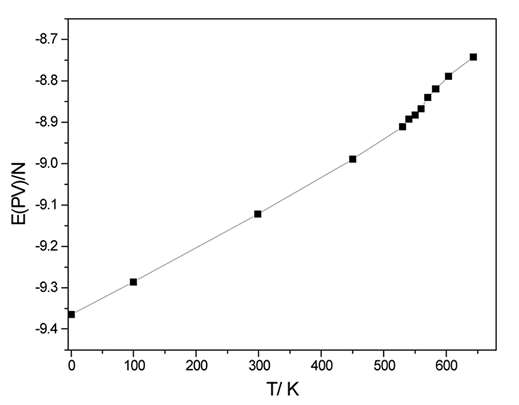
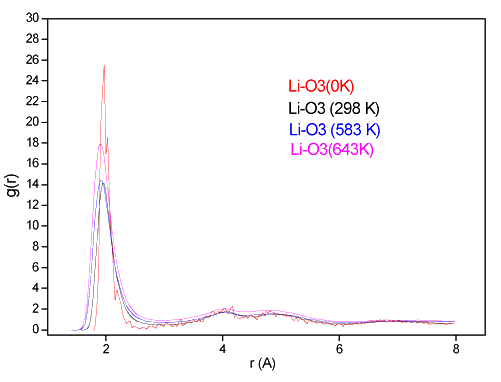
Fig. 1.
Entalpia (eV) X T (K) Fig.2. RDF para
Li- O
Os
coeficientes de difusão na fase líquida estão em
torno de 3x10-9 m2s-1, e concordam
com resultados experimentais disponíveis para carbonatos de
lítio8.
O
modelo proposto para uma fase cristalina do acetato de lítio
anidro apresentou densidade de 1,20 g/cm3 a 0 K, em
razoável concordância com o valor experimental de 1,23
g/cm3.
Os
resultados obtidos permitiram propor um modelo para as fases
cristalinas e líquida do acetato de litio, e estão
sendo aplicados ao estudo da estrutura de materiais com baixo ponto
de fusão, úteis à incorporação de
compostos orgânicos com propriedades ópticas especiais.
Mort, K.
A., Tese de doutorado,1997, University of Liverpool, UK.
Mcbride,
C. e Wilson, M. R., Molecular Physics, 1999, 97 (4), 511-522.
Barreto,
L. S. e Alves, O.L., 1998, CBECIMAT , Curitiba, Paraná.
Barreto,L.S.
e Alves,O.L.,1998,21aReunião Anual da SBQ,Poços
de Caldas, MG.
Forester,
T. R., Smith, W., 1998, DLPOLY: a molecular dynamics program, CCP5
Program Library (Manchester, UK:Daresbury Laboratory)
Sounderson,
C.P. , Tese de mestrado, 1960, University of Manitoba, Winnipeg,
Canada.
Barreto,
L. S., Mort, K. A., Jackson, R.A., Alves, O. L., 1999, CCP5 - Annual
Meeting, Birmingham University, UK.
Tissen,
J. T. W. and Janssen, G. J.,1990, Molecular Physics, 71(2),
413-426.
[CAPES/PICDT/PDEE]