QUIMIOMETRIA APLICADA A ESTUDOS CONFORMACIONAIS
Aline Thaís Bruni (PG) e Márcia M. C. Ferreira (P.Q.)
Instituto de Química, Universidade Estadual de Campinas (Unicamp)
palavras-chave: quimiometria, análise conformacional, fármacos
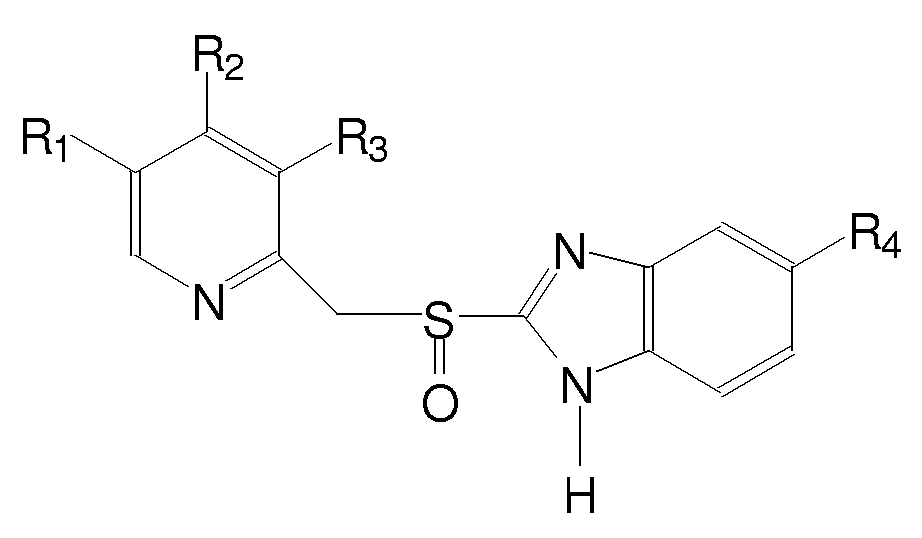
Este trabalho tem por objetivo principal aplicar técnicas de quimiometria a cálculos teóricos para a determinação da conformação mais estável de três fármacos: omeprazol, pantoprazol e lansoprazol.(1) Estes fármacos são benzimidazóis substituídos que suprimem a secreção ácido-gástrica por inibição da enzima H+, K+- ATPase (Figura 1).
Sistemas com muitos graus de liberdade têm comumente propriedades dinâmicas e termodinâmicas determinadas pela natureza das superfícies de energia potencial. A energia eletrônica mais baixa de uma molécula depende somente das coordenadas nucleares. A análise conformacional é então de extrema importância porque muitas propriedades moleculares dependem criticamente das estruturas tri-dimensionais. Análises dos espaços conformacionais moleculares são utilizados para localizar estruturas estáveis de vários tipos de moléculas.(2)
Existem várias técnicas que podem ser utilizadas para achar mínimos de energia.(3) Aqui foi utilizada a técnica chamada análise conformacional sistemática. A grande vantagem desta técnica é que há um ponto final definido na superfície potencial. Contudo, a grande desvantagem é o fenômeno denominado explosão combinatória. Assim, para resolver este problema, foi introduzido neste trabalho uma nova metodologia.
|
Posição |
Omeprazol |
Pantoprazol |
Lansoprazol |
|
R1 |
CH3 |
- |
- |
|
R2 |
OCH3 |
OCH3 |
OCH2CF3 |
|
R3 |
CH3 |
OCH3 |
CH3 |
|
R4 |
OCH3 |
OCF2H |
- |
Figura1 .Estrutura básica dos fármacos estudados
Esta metodologia utiliza a técnica de Análise de Componentes Principais (PCA) juntamente com cálculos de química quântica para estudar a superfície de energia potencial. Através desta técnica é possível resolver o problema da explosão combinatória associada à análise conformacional sistemática por diminuição da dimensão dos sistemas estudados. Neste trabalho, as estruturas iniciais dos fármacos em questão foram montadas pelo programa Spartan. Para conduzir a busca conformacional, as configurações foram geradas girando-se sistematicamente os diedros envolvendo as rotações livres indicadas pelas setas na estrutura básica dos fármacos (Figura 1). Inicialmente o incremento de ângulo utilizado foi de 30°. Estas rotações foram feitas em pares entre as ligações envolvidas, e para cada par obteve-se uma superfície de energia potencial. Os cálculos foram realizados pelo método semi-empírico AM1, implementado no programa Gaussian 94. As matrizes de dados relativas às superfícies em questão foram submetidas em conjunto à PCA e as regiões de energia mínima foram separadas. Os cálculos foram refinados nestas regiões, com um incremento de ângulo de 5°. Novamente estes dados foram submetidos à PCA, e então foram separadas as conformações de menor energia para os fármacos em questão. A Tabela 1 mostra os resultados obtidos para cada um dos compostos estudados. Tem-se que para o pantoprazol, foram obtidas duas estruturas de mínima energia, enquanto que para o omeprazol e para o lansoprazol foram obtidas quatros estruturas de mínima energia. Todas as estruturas de mínimo apresentam valores de energia muito próximos.
|
Tabela 1: Valores de energia/Kcal.mol-1 |
|||||
|
Pantoprazol |
Omeprazol |
Lansoprazol |
|||
|
Estrutura |
Energia |
Estrutura |
Energia |
Estrutura |
Energia |
|
1 |
-100.27 |
1 |
-36.18 |
1 |
-78.86 |
|
2 |
-100.99 |
2 |
-35.67 |
2 |
-77.61 |
|
|
|
3 |
-35.16 |
3 |
-79.24 |
|
|
|
4 |
-34.79 |
4 |
-78.92 |
Assim, conclui-se que a técnica quimiométrica de Análise de Componentes Principais aplicada a cálculos teóricos foi eficiente e útil não somente para reduzir a dimensão dos dados dos sistemas estudados, como também para encontrar as regiões de mínimos globais nas superfícies estudadas.
Bibliografia
Lindberg, P.; Nordberg, P.; Alminger, T., Brändströn, A.; Wallmark, B.; Journal of Medicinal Chemistry, 29,1329 (1986). Tanaka,M.; Yamazaki, H., Hakusui, H.; Nakamichi, N.; Sekino, H.; Chirality,9,17 (1997). Landes,B.D.;Petite,J.D.;Flouvat,B.; Clinical Pharmacokinetics, 28,1158 (1995).
Jensen, F.; Introduction to Computational Chemistry, John Wiley & Sons, 1999.
Leach, A. R.; Molecular Modelling, Longman Singapore Publishers Ltc., 1996.