Os cálculos foram realizados em nível RHF com base 631G com polarização simples em todos os átomos utilizando o programa GAMESS [4] versão dezembro de 1999 em um cluster de computadores Pentium II rodando sistema operacional FreeBSD 3.4.
Estudo Ab Initio da Primeira Camada de Solvatação do Íon Esquarato
Lucimara Ramos Martins (PG), Munir Salomão Skaf (PQ) , Pedro A.M.Vazquez (PQ)
Departamento de Físico-Química
Instituto de Química – Universidade Estadual de Campinas
Palavras-Chave: dinâmica molecular, oxoânions, ab initio
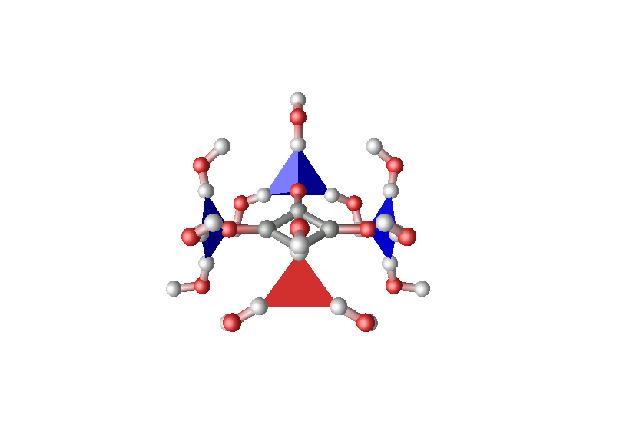
Analisando as funções de distribuição de pares dos sítios H e O da água ao redor dos sítios C e O do íon obtidas nas simulações, construímos uma estrutura para a primeira camada de solvatação. As moléculas de água ao redor dos oxigênios do íon oxocarbono se encontram dispostas nos vértices de uma pirâmide. Os três hidrogênios ao redor de um dos oxigênios do ânion se encontram nos vértices de um triângulo equilátero disposto perpendicularmente ao plano do íon a uma distância de 0,82 Å. Os ângulos HOoxoC são todos 120 graus e os ângulos diedros H-Ooxo-C-C são –90, 30 e 150 graus. Os oxigênios das moléculas de água se encontram nos vértices de um triângulo equilátero semelhante ao primeiro a uma distância de 1,02 Å do oxigênio do íon. Os vértices de pirâmides situadas em oxigênios vizinhos no íon são alternados o que permite a interação entre as moléculas de água dos ângulos diedros de 30 e 150 graus com aquelas localizadas nos ângulos diedros de 90 graus ao redor dos oxigênios vizinhos (Figura).
O objetivo deste trabalho é, portanto, comprovar através de cálculos quânticos a estabilidade da estrutura proposta para a primeira camada de solvatação construída a partir das funções de distribuição de pares determinadas por simulação de dinâmica molecular.
Os
cálculos foram realizados em nível RHF com base 631G
com polarização simples em todos os átomos
utilizando o programa GAMESS [4] versão dezembro de 1999 em um
cluster de computadores Pentium II rodando sistema operacional
FreeBSD 3.4.
Os hidrogênios das moléculas de água que estão mais afastados do íon foram orientados de tal forma a não formarem ligações de hidrogênio com as águas vizinhas. Como resultado, obtivemos uma estrutura na qual as águas localizadas nos diedros de 30 graus fazem ligações de hidrogênio com as águas localizadas nos diedros de –150 graus da carbonila vizinha e as moléculas de água localizadas nos diedros de 90 e –90 graus não fazem ligações de hidrogênio com nenhuma molécula de água. Para verificar se a estrutura proposta era menos estável do que a obtida no cálculo quântico, realizamos a mesma otimização após reorientarmos as águas para realizarem as ligações de hidrogênio propostas na estrutura determinada através das funções de distribuição de pares.
O resultado obtido mostra uma estrutura otimizada com poucas alterações em relação a estrutura proposta: os ângulos diedros são alterados de 150 para 147 graus e de 30 para 33, as águas nos diedros de 90 graus são levemente aproximadas e as demais afastadas. Em termos energéticos, a segunda estrutura possui uma energia um pouco menor. As cargas de Mulliken sobre os átomos de oxigênio praticamente não foram alteradas em relação aos cálculos efetuados com a mesma base para o íon no estado gasoso (cargas utilizadas sobre os sítios de O do íon nas simulações de dinâmica).
G.Seitz, Imming, Chem Rev., 92, 1256 (1992)
M.C.C. Ribeiro, L.F.C. de Oliveira, P.S. Santos, Chem. Phys., 217, 71 (1997)
L.R.Martins, M.S.Skaf, X Simpósio Brasileiro de Química Teórica, Caxambu, MG, Nov. 1999, p-117.
M.W. Schmidt, K.K. Baldridge, J.A.Boatz, S.T.Elbert, M.S.Gordon, J.H. Jensen, S.Koseki, N.Matsunaga, K.A. Nguyen, S.J.Su, T.L.Windus, M.Dupuis, J.A.Montgomery, J. Comput. Chem.14, 1347-1363 (1993).
FAPESP