ANÁLISE
CONFORMACIONAL DE UMA DIAMIDA DA ARGININA, ENVOLVENDO CÁLCULOS
DE SOLVATAÇÃO EM ÁGUA.
Maria
Cristina Andreazza Costa (PQ)1, Yuji Takahata (PQ)2
e
Luiz Carlos Gomide Freitas (PQ)1.
1
Departamento de Química, Universidade Federal de São
Carlos
2
Departamento de Físico Química, Instituto de Química,
UNICAMP
Palavras-chave:
neolignanas, arginina, leishmania
INTRODUÇÃO
O
planejamento de fármacos, utilizando química
computacional, requer uma análise conformacional acurada, uma
vez que este é o ponto de partida para a correlação
com a atividade biológica. É desejável a
utilização de uma metodologia que forneça
resultados não só de cálculos no vácuo,
mas num sistema que possa representar melhor o meio biológico.
Um
bom modelo para esse tipo de análise, talvez seja o da
solvatação, tanto da droga quanto do receptor, em água.
Neste trabalho, estamos focalizando um possível receptor às
drogas anti-leishmaniose: Uma diamida da arginina.
Cálculos
anteriores, no vácuo (apresentados na SBQT/99), nos mostraram
a presença de 24 mínimos locais para a diamida em
questão. Para continuar os estudos de interação
fármaco-receptor, devemos procurar aquele que melhor se ajuste
ao nosso principal objeto de estudo: Neolignanas ativas em
leishmaniose.
Podemos
estudar sistemas como este que apresentamos (uma diamida solvatada
por moléculas de água), pelo cálculo de
propriedades termodinâmicas de equilíbrio; para isso,
escolhemos o método de Monte Carlo [1].
METODOLOGIA
Os
24 mínimos locais encontrados anteriormente, com o método
semi-empírico MNDO-PM3, foram reotimizados com o método
ab initio e conjunto de base 6-31G**, através do
programa Gaussian; estes cálculos foram realizados no CENAPAD.
As
novas geometrias obtidas, foram então solvatadas por moléculas
de água, com o método de Monte Carlo e o programa
Diadorim [2]. Utilizamos uma caixa cúbica contendo 800
moléculas de água, à temperatura biológica
(36,5o) e pressão de 1,0 atm; o modelo TIP4P foi
escolhido para representar a água. As energias de interação
soluto-solvente (neste caso, diamida-água), nos deram
informações sobre os mínimos locais mais
estáveis em água.
RESULTADOS
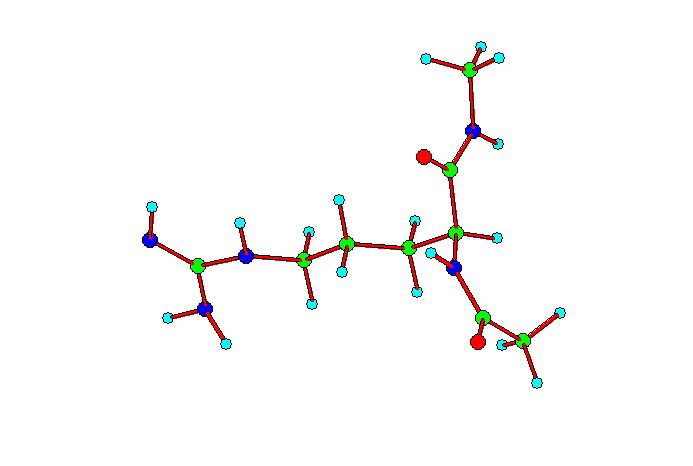
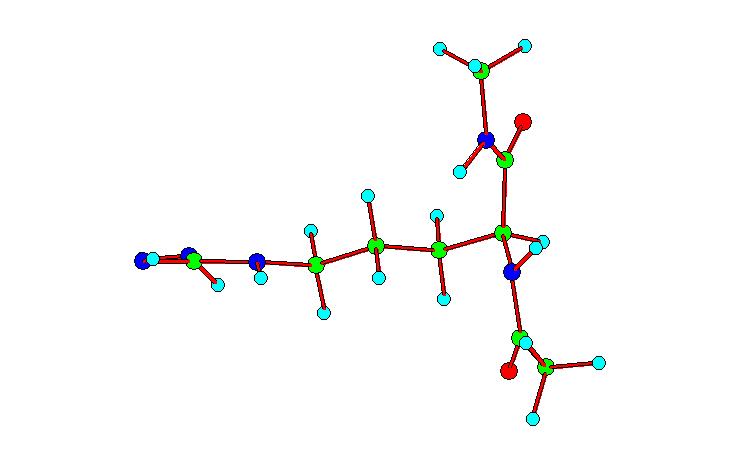
Após
a reotimização das geometrias da diamida com o método
ab initio , verificamos que, ao invés de 24 mínimos
locais, temos agora 20 conformações distintas.
Observamos dois “tipos” principais de conformações:
1) Aqueles em que o grupo guanidínico da diamida da arginina
comparece no mesmo plano que as amidas; 2) Aqueles que apresentam os
dois grupos citados, perpendiculares entre si; dentro destes dois
tipos, encontramos uma variedade de conformações, com
as diamidas estendidas ou dobradas.
Tipo 1
Tipo 2
Um
trabalho realizado anteriormente (SBQT/99), mostrou que as
conformações do tipo 1 sobrepõem-se melhor às
neolignanas, do que do tipo 2 ou daquelas em que as diamidas estão
dobradas. Observamos cerca de dez conformações do tipo
1 e quatro delas já foram solvatadas em água
(conformações A, B, C, D). Os seguintes valores de
energia foram obtidos, pelas análises realizadas no vácuo
(energia em u.a) e em água (energia de interação
soluto-solvente, em kcal/mol): A(-773,92081 u.a. e –85,55
kcal/mol); B ( -773,92631 u.a e –80,46 kcal/mol); C
(-773,92633 u.a. e –79,33 kcal/mol); D (-773,92624 u.a. e
–84,52 kcal/mol). A ordem de estabilidade encontrada pelos
cálculos no vácuo foi totalmente inversa à ordem
obtida em água; no vácuo, observou-se: EC <
EB < ED < EA
e as energias de interação soluto-sovente apresentaram
a seguinte ordem: EA < ED<
EB < EC.
Concluímos que a solvatação em água
influencia bastante os resultados da análise conformacional e
continuaremos essa análise até encontrarmos o mínimo
local mais adequado.
Bibliografia
[1]
Allen, M.P., Tildesley, D., “Computes Simulation of Liquids”,
Clarendon Press, Oxford (1987).
[2]
Programa Diadorim, escrito em linguagem Fortran por Luiz Carlos
Gomide Freitas, Departamento de Química, UFSCar, 1992.
FAPESP, CNPq