MONTE
CARLO VARIACIONAL: UMA ABORDAGEM ATRAVÉS DA TEORIA DE MATRIZ
DENSIDADE
José
Roberto dos Santos Politi (PG) e Rogério Custodio (PQ)
Universidade
Estadual de Campinas UNICAMP
Instituto
de Química Departamento de Físico-Química
palavras-chave:
estrutura eletrônica, monte carlo quântico e amostragem
preferencial
O
método Monte Carlo (MC) é uma técnica
estocástica que permite o cálculo de integrais,
particularmente eficiente para funções com múltiplas
variáveis [1].
Recentemente, o MC tem sido empregado
na simulação de sistemas quânticos, denominado
Monte Carlo Quântico (MCQ). O MCQ
mais simples é o Monte Carlo Variacional (MCV), que
utiliza o algoritmo de Metropolis [2]
e o princípio variacional para calcular o valor esperado da
energia (E) (eq1) e outras
propriedades associadas à função de onda
tentativa y.
(1)
O
peso estatístico é dado pela função
densidade de probabilidade de elétrons:
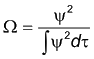 . Logo:
. Logo:
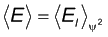 .
O subscrito y2 indica
que esta é uma média ponderada por W.
A energia local (El) é calculada como a
soma da energia cinética local (Tl) e a
energia potencial local (Pl). A Tl
é descrita por um operador diferencial que não permite
o cancelamento da função de onda, mais especificamente
das funções de spin, o que inviabiliza o cálculo.
Para obter um valor numérico para Tl, têm-se
utilizado uma função de onda teste [3] que fatora os
elétrons de spins opostos em um produto de dois determinantes
de Slater :
.
O subscrito y2 indica
que esta é uma média ponderada por W.
A energia local (El) é calculada como a
soma da energia cinética local (Tl) e a
energia potencial local (Pl). A Tl
é descrita por um operador diferencial que não permite
o cancelamento da função de onda, mais especificamente
das funções de spin, o que inviabiliza o cálculo.
Para obter um valor numérico para Tl, têm-se
utilizado uma função de onda teste [3] que fatora os
elétrons de spins opostos em um produto de dois determinantes
de Slater :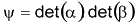 .
Assim, é obtida a independência de Tl
com as coordenadas de spin. Este tratamento preserva algumas
características da função de onda original, como
os nós da função e a condição de
cúspide. Entretanto, outros aspectos importantes do
determinante de Slater monoeletrônico são violados, como
a antisimetria e a indistinguibilidade eletrônica. A
alternativa que apresentamos para resgatar a integridade e o
formalismo da química quântica é reescrever a El
em termos da matriz densidade de ordem n:
.
Assim, é obtida a independência de Tl
com as coordenadas de spin. Este tratamento preserva algumas
características da função de onda original, como
os nós da função e a condição de
cúspide. Entretanto, outros aspectos importantes do
determinante de Slater monoeletrônico são violados, como
a antisimetria e a indistinguibilidade eletrônica. A
alternativa que apresentamos para resgatar a integridade e o
formalismo da química quântica é reescrever a El
em termos da matriz densidade de ordem n:
(2)
sendo
x as coordenadas espaciais e x a
coordenada de spin. Assim, a Tl e a El
podem ser calculadas, independentes das funções de
spin. Para reduzir a taxa de rejeição de configurações,
foi incorporada a amostragem preferencial. Desta forma, os
deslocamentos aleatórios são direcionados por r
(eq.3), gerando configurações significativas para a
média [4]:
 (3)
(3)
y
é a nova coordenada, x é a coordenada inicial,
D=0,5, dt é o
intervalo de tempo imaginário para o deslocamento, c
é uma variável randômica obtida por uma
distribuição gaussiana e F é definido
pela equação abaixo:
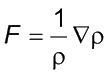 (4)
(4)
Utilizando
esta metodologia, apresentamos resultados para o átomo de
lítio, investigando a convergência dos resultados em
função da taxa de aceitação. Os
resultados obtidos concordam com a literatura, comprovando a
viabilidade da metodologia. A taxa de aceitação que
apresenta a melhor convergência se encontra entre 50% e 70%.
|
Tabela
1 Energia do átomo de lítio (u.a.), utilizando
função single-zeta [5].
|
|
|
|
|
|
|
|
|
|
|
Aceitação
(8x106 configurações)EnergiaDesvio
Padrão20%-7,4179600,00051250%-7,4182930,00053170%-7,4188980,000544Clementi
[5]-7,4184820M.H. Kalos and P.A. Whitlock, Monte Carlo Methods.
(Wiley,NY,1986)
N.
Metropolis, et alli J.Chem.Phys. 21 (6), 1028
(1953)
P.J.
Reynolds, D.M. Ceperley, B.J. Alder, W.A. Lester Jr., J. Chem.
Phys. 77(11), 5593(1982)
B.L.
Hammond, W.A. Lester Jr and P.J. Reynolds Monte Carlo Methods in
Ab Initio Quantum Chemistry (World Scientific,Singapore,1994)
E.
Clementi, C. Roetti; Atomic Data and Nuclear Data Tables
14(3-4),177
(1974) (CNPq,FAPESP,FAEP,CENAPAD-SP)