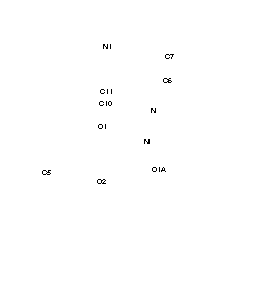
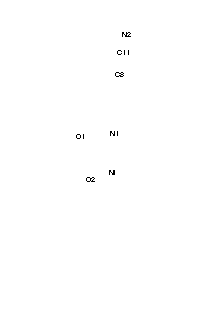
Crystal structures of adducts of Ni (II) acetylacetonate (AcAc) chelate with heterocyclic bases.
J. Zukerman-Schpectora,b (PQ), Antonio Carlos Trindadea (PG) and
Pedro O. Dunstanc (PQ)
aDepartamento de Química Fundamental, Instituto de Química, USP; bLaboratório de Cristalografia, Estereodinâmica e Modelagem Molecular, Departamento de Química, UFSCar; cInstituto de Química, UNICAMP.
Palavras chave: Ni(II)actilacetonato, difração de raios-X, estereoquímica
Adducts of Ni (II) acetylacetonate chelated with heterocyclic bases, such as quinoline, pyridine, 4-cyanopyridine, morpholine and 3-metylpyridine, have been synthesized aiming at studying the conformational rigidity induced by the different ligands and therefore determining the distortions in the six-coordinated complexes. In this work we report two crystal structures of the Ni(II)(AcAc)2 .2L, with L = 3 and 4-cyanopyridine. The adducts were synthesized by the reaction of the Ni (II) acetylacetonate and ligands in methanol, the solids were then washed with petroleum ether and dried in vacuo. Crystals suitable for X-ray analyses were obtained from methanol, by slow evaporation at room temperature. Crystal data were measured on an Enraf-Nonius Cad-4 diffractometer using the q/2q scan technique, at room temperature, and graphite monochromated MoKa radiation. Cell parameters were refined by least-squares-method on the basis of 25, high angle, independent reflections. Data were collected up to q = 25.5° and corrected for Lorentz-polarization and absorption (y-scan) effects. Three standard reflections were measured throughout the experiment and showed no significant variation. The structures were solved by the heavy-atom Patterson method (SHELXS86[1]) and difference Fourier syntheses and refined anisotropically by full-matrix least-squares on F2 (SHELXL97[2]). H-atoms were located on stereochemical grounds and refined with fixed geometry, each riding on a carrier atom, with an isotropic displacement parameter equal to 1.5 (for methyl H atoms) or 1.2 (for the other H atoms) times the values of the equivalent isotropic displacement parameter of the atom to which they were attached. Final difference Fourier maps showed no peaks of the chemical significance. Drawings were done using ZORTEP[3]
(1)Crystal data for the 3-cyanopyridine complex: Ni(II)(C22H22N4O4), (blue), fw=465.14, monoclinic, P21/a, a = 7.8295(6), b = 19.3334(18), c = 8.1244(10) Å, b=116.019(8)o , V=1105.16(19) Å3, Z=2, S=0.953, R1=0.0450 for 1131 reflections with I.>2s(I) and 144 parameters refined.
(2)Crystal data for the 4-cyanopyridine complex: Ni(II)(C22H22N4O4), (blue), fw=465.14, triclinic, P-1, a = 6.2795(6), b =9.912(1), c =10.392(1) Å, a=64.96(1), b=86.441(8) , g=73.333(9)o, V=560.1(6) Å3, Z=2, S=1.055, R1=0.0353 for 1605 reflections with I.>2s(I) and 144 parameters refined.
(2)
Discussion: In both compounds the AcAc moiety is planar, to within experimental accuracy, the r.m.s deviation of the seven atoms being of 0.012 and 0.018 for (1) and (2) respectively. The nickel atoms does not lie in the plane of the AcAc residue but 0.247(4) and 0.145(3) Å from it, for (1) and (2) respectively. Ni (II) is sited on a center of symmetry and is octahedrically bonded to two equatorial AcAc groups and two 3-cyanopyridine (1) and two 4-cyanopyridine (2), that are axially coordinated in a trans configuration. The Ni-OAcAc distances in (2) of 2.0110(16) and 2.0238(18) Å give rise to the called tetragonal distortion, being this distortion much less in (1) in which the distances are 2.009(2) and 2.016(2) Å. In the solid state no short intramolecular distances were found. The shortest intermolecular distances found for the N-atom of the cyano group and C atoms of the AcAc moiety are: (1) N2...H1Ai = 3.832(6) Å, N2...H1Ai-C1i = 164o; (2) N2...H5Bii = 2.75, N2...C5ii = 3.607(5) Å, N2...H5Bii = 149º Moreover, there is another interaction in (1) with a phenyl H-atom:N2...H9iii = 2.79, N2...C9iii = 3.503(6) Å, N2...C9-H9iii = 134o (i = 1.5 - x, -0.5 + y, 1 - z, ii = -x, 1 - y, -1 - z, iii = -0.5 + x, 0.5 - y, -1 + z).To whether these interactions are hydrogen bonds it is difficult to assert because as has been pointed out "the field is getting mudier and mudier as the definition of a hydrogen bond is relaxed".In any case in (2) the N...H distance is marginally longer than the sum of the Van der Waals radii of H and N (2.7 Å)
[1] Sheldrick, G.M. SHELXS86 (1990). Acta Cryst. A46, 467.
[2]. Sheldrick, G.M (1997) SHELXL97. Program for the Refinement of Crystal Structures. University of Götingen, Germany.
[3] Zsolnay, L.(1995); ZORTEP. An interactive molecular graphics program. University of Heidelberg, Germany.
FAPESP, CNPq