HIDRÓLISE BÁSICA DE ÉSTERES: UM ESTUDO AB INITIO DA SUPERFÍCIE DE ENERGIA POTENCIAL PARA A REAÇÃO HCOOCH3 + OH- EM FASE GASOSA
Josefredo R. Pliego Jr. (PQ) e Jose M. Riveros (PQ)
Instituto de Química – Universidade de São Paulo
Palavras-chave: mecanismo de reação, cálculos ab initio, adição a carbonila
A hidrólise básica de ésteres é uma das reações mais conhecidas da química ôrganica, e tem grande importância em química e bioquímica. Vários estudos experimentais apontam que esta reação ocorre em fase aquosa pelo mecanismo BAC2, ou seja, adição do íon hidróxido à carbonila, formando um intermediário tetraédrico, seguido pela eliminação de álcool e formação do carboxilato correpondente. Entretanto, este padrão de reatividade não é intrinseco às moléculas de ésteres, sendo na realidade fortemente influenciado pelo solvente. De fato, estudos da reação de ésteres com hidroxila em fase gasosa mostram que outros caminhos podem atuar1. No caso do formiato de metila, a reação pode proceder por substituição nucleofílica, gerando os mesmos produtos, ou por descarbonilação, onde o hidrogênio ligado a carbonila é abstraído pelo íon OH-, formando água, metóxido e monóxido de carbono.
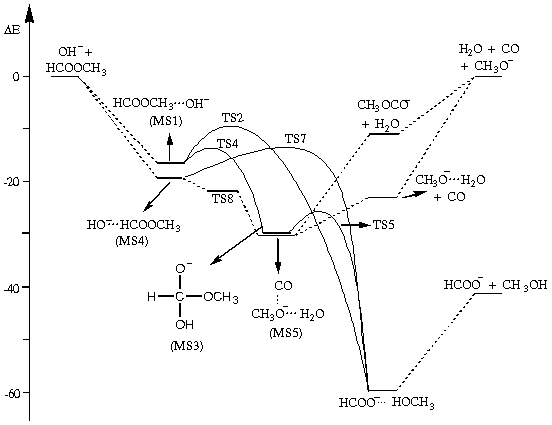
Apesar da importância desta reação, nenhum estudo teórico deste tipo de reação em fase líquida foi reportado, e apenas dois estudos em fase gasosa foram feitos2. No presente trabalho, nós reportamos um estudo ab initio da reação do íon hidroxila com formiato de metila em fase gasosa. As geometrias de equilíbrio, estados de transição e frequências harmônicas foram determinadas em nível MP2/6-31+G*. Cálculos da energia no ponto foram realizados em mais alto nível de correlação eletrônica (MP4) e estendendo a base para 6-311+G(2df,2p). Os resultados estão sumarizados na figura 1.
A aproximação do íon hidroxila do formiato de metila leva a duas estruturas preferenciais: MS1 e MS4, os quais são complexos do tipo íon-dipolo. Entretanto, a estrutura MS4 não sobrevive como mínimo quando a base e o nível de correlação são estendidos, de modo que a aproximação do hidroxila desta posição na molécula deve levar diretamente a MS5. O caminho a ser seguido por este complexo é a perda de CO e formação do complexo água-metóxido. No caso dos reagentes terem excesso de energia, a dissociação de MS5 pode ser completa, levando a monóxido de carbono, água e metóxido, o que de fato é observado experimentalmente. O complexo MS1 pode se rearranjar pelos caminhos envolvendo os estados de transição TS2 e TS4. O caminho através do TS2 corresponde a um mecanismo Sn2, enquanto através do TS4 envolve o intermediário tetraédrico. Nossos cálculos sugerem que TS4 é o caminho preferencial, o que também é consistente com as observações experimentais. Em suma, o presente estudo apresenta uma visão geral da superfície de energia potencial para esta reação em fase gasosa, e racionaliza os dados experimentais. O estudo desta reação em fase líquida está em andamento.
Figura 1
1 - (a) Faigle, J. F. G.; Isolani, P. C.; Riveros, J. M. J. Am. Chem. Soc. 1976, 98, 2049. (b) Isolani, P. C.; Riveros, J. M. Chem. Phys. Lett. 1975, 33, 362. (c) Takashima, K.; Riveros, J. M. J. Am. Chem. Soc. 1978, 100, 6128.
2 - (a) Pranata, J. J. Phys. Chem. 1994, 98, 1180. (b) Jorgensen, W. L.; Blake, J. F.; Madura, J. D.; Wierschke, S. D. A. C. S. Symp. Ser. 1987, 353, 200.