|
(0,008mM) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ANÁLISE CONFORMACIONAL DE DERIVADOS DE OXAZOLA: AGONISTAS NÃO PROSTANÓIDES DO RECEPTOR DE PGI2
Rita Cristina Azevedo Martins (PG) e-mail rmatins@.iq.ufrj.br
Ricardo Bicca de Alencastro (PQ) e Magaly Girão Albuquerque (PQ)
Laboratório de Modelagem Molecular (LabMMol) – Departamento de Química Orgânica - Instituto de Química - Universidade Federal do Rio de Janeiro
palavras-chave: oxazola, prostaciclina, semi-empírico
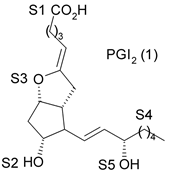
1. Introdução A prostaciclina I2 (PGI2) (1) (Fig. 1) é um dos mais potentes vasodilatadores e inibidores da agregação plaquetária1,2 e é encontrada em tecidos vasculares e plaquetas sanguíneas1. Entretanto, a PGI2 não é empregada como agente terapêutico porque tem tempo de meia-vida de 3 min, aproximadamente, em condições fisiológicas3,4. No desenvolvimento de novos agentes antiagregante- plaquetários com melhor perfil farmacocinético, Meanwell et al.5,6,7 sintetizaram e avaliaram a atividade biológica de derivados de oxazola (ácidos 4,5-difenil-2-oxazola e 4,5-difenil-5-oxazolil-2-oxazola)7,9,10 (2-12) (Fig. 1) denominados de prostaciclina miméticos não prostanóides por serem estruturalmente distintos da PGI2. Na literatura encontram-se modelos de sítios farmacofóricos para agonistas do receptor de PGI2 (IP),7,9 entretanto estes modelos bidimensionais não são capazes de definir com precisão a disposição espacial dos grupamentos farmacofóricos.
2. Objetivo Na falta da estrutura tridimensional do receptor IP, a abordagem mais racional é o estudo do agonista natural e de agonistas e antagonistas sintéticos por técnicas de química computacional11. O objetivo deste trabalho é definir as características topográficas comuns à PGI2 (1) e aos agonistas do receptor IP que podem ser correlacionadas com a atividade biológica, através do estudo conformacional de derivados oxazola não prostanóides (2-12) do receptor IP, utilizando-se métodos teóricos de mecânica molecular e de mecânica quântica à nível semi-emprírico.
3. Metodologia As etapas de construção, otimização da geometria, minimização da energia das estruturas e análise conformacional foram realizadas no programa PC Spartan Pro v. 1.0.12 Nos cálculos de mecânica molecular utilizou-se o campo de força Merck Molecular Force Field (MMFF)13 e nos cálculos semi-empíricos, o Hamiltoniano Austin Model 1 (AM1)14. A etapa de sobreposição foi realizada no programa Insight II 15 numa estação de trabalho Silicon Graphics O2.
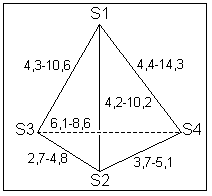
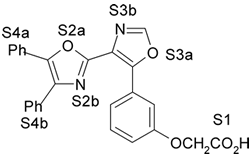
4. Resultados e Discussão Os grupos farmacofóricos dos derivados de oxazola foram definidos, de acordo com a estrutura da PGI2, como S1 a S4 (Fig. 1): S1 representa um sítio ácido que pode estar ionizado no meio biológico, interagindo com o receptor por ligação iônica, ou na sua forma não ionizada, por ligação hidrogênio; S2 representa um sítio de ligação hidrogênio, S3 representa um grupo espaçador ou sítio de ligação hidrogênio, e S4 representa um sítio hidrofóbico. As distâncias entre os sítios farmacofóricos das conformações obtidas para a PGI2 e os compostos mais ativos (1-7) compõem o modelo farmacofórico apresentado na Figura 2. A análise mostra que as distâncias entre os grupos farmacofóricos dos compostos menos ativos (8-12) estão dentro da faixa definida no modelo obtido (Fig. 2). Entretanto, a análise de sobreposição molecular entre a PGI2 (1) e os derivados de oxazola (2-12) mostra que o sítio S4 não se sobrepõe perfeitamente ao sítio correspondente da PGI2. A diferença de atividade entre os compostos da série estudada não pode ser justificada pelas distâncias entre os grupos farmacofóricos, mas pela orientação dos mesmos no espaço. Adicionalmente, o caráter e o volume do substituinte no sítio S3 ou próximo a ele, também estão implicados na variação da atividade, visto que os compostos mais ativos tem um heteroátomo aceptor de ligação hidrogênio ou um grupo espaçador. O composto 12, por exemplo, apesar do heteroátomo em S3, contém um substituinte fenila tendo sua atividade reduzida.
|
(0,008mM) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 1. Estrutura dos compostos (1-12), definição dos sítios farmacofóricos (S1 a S4) e IC50 (mM).
|
Figura 2. Modelo de distâncias (Å) entre os sítios farmacofóricos de 1-7. |
5. Conclusões O modelo de distâncias entre os grupos farmacofóricos obtido neste trabalho não é capaz de justificar a variação de atividade dos compostos estudados, entretanto, a análise de sobreposição molecular evidencia que não há sobreposicão simultânea de todos os grupos farmacofóricos levando a uma menor interação dos compostos no sítio receptor da PGI2. |
6. Referências Bibliográficas
|
1-Vane, et al., 1995 Am. J. Cardiol., 75 (19), 3A. |
19-aMeanwell, et al., 1993 J. Med. Chem., 36 3871. |
|
2-Collins, et al 1993 Chem. Rev., 93, (4) 1533. |
10-bMeanwell, et al., 1993 J. Med. Chem., 36 3884. |
|
3-Campbell 1990 In Goodman & Gilman’s, 24. 8th ed. |
11-Barreiro, et al., 1997 Química Nova, 20 300. |
|
4-Ullrich, et al 1994 Ang. C. Int. Ed Engl., (33) 1991. |
12-PC Spartan Pro 1.0 1999, Wavefunction, Inc. |
|
5- aMeanwell, et al.,1992 J. Med. Chem., 35 389 |
13-Halgren, et al., 1996 J. Am. Chem. Soc., 99, 4899. |
|
6-bMeanwell, et al., 1992 J. Med. Chem., 35 3483. |
14-Dewar, et al 1985 J. Am. Chem. Soc., 107 3902. |
|
7-cMeanwell, et al., 1992 J. Med. Chem., 35 3498 |
15-INSIGHT II 1995 Molecular Simulations Inc. |
|
8-Patani, et al., 1996 Chem. Rev., 96, 3147. |
Agradecimentos: CAPES, CNPq, FAPERJ E FUJB |
 2
(0,025mM)
2
(0,025mM)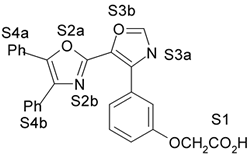 3
(0,16mM)
3
(0,16mM)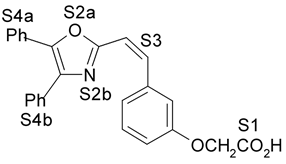 4
(0,18mM)
4
(0,18mM)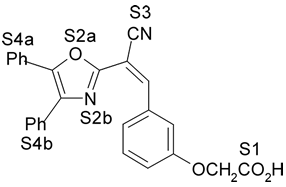 5
(0,25mM)
5
(0,25mM)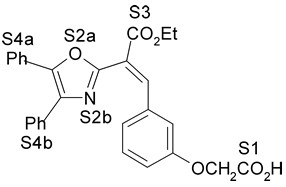 6
(0,36mM)
6
(0,36mM)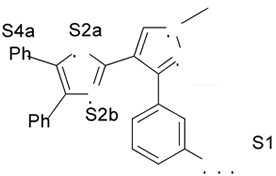 7
(0,65mM)
7
(0,65mM)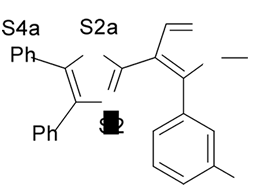 8
(1,06mM)
8
(1,06mM)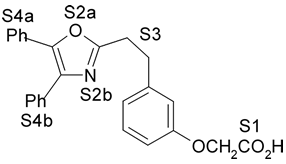 9
(1,2mM)
9
(1,2mM)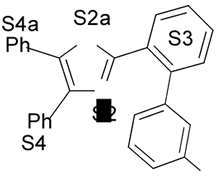 10
(11,1mM)
10
(11,1mM)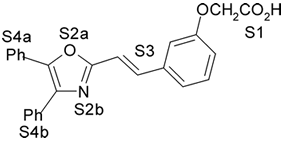 11
(13,0mM)
11
(13,0mM)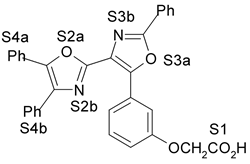 12
(>60mM)
12
(>60mM)